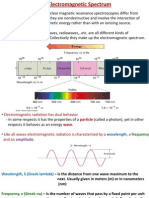
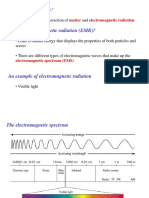
You might also like
- NMR Case Studies: Data Analysis of Complicated MoleculesFrom EverandNMR Case Studies: Data Analysis of Complicated MoleculesRating: 5 out of 5 stars5/5 (1)
- Theoryofirspectroscopy 160622080111Document50 pagesTheoryofirspectroscopy 160622080111ArshNo ratings yet
- Chapter3 IR1Document34 pagesChapter3 IR1dokdangNo ratings yet
- Atomic Absorption Spec.Document65 pagesAtomic Absorption Spec.Donald100% (1)
- FALLSEM2021-22 BCHY101L TH VL2021220106627 Reference Material I 24-12-2021 EC Module 6 - MARDocument66 pagesFALLSEM2021-22 BCHY101L TH VL2021220106627 Reference Material I 24-12-2021 EC Module 6 - MARHarsh AgarwalNo ratings yet
- CHM556/ CHM557 Organic Chemistry Ii/ Organic Chemistry: Spectroscopy of Carbon CompoundsDocument200 pagesCHM556/ CHM557 Organic Chemistry Ii/ Organic Chemistry: Spectroscopy of Carbon CompoundsnanaNo ratings yet
- Chapter 1 Full Chm556 2018Document206 pagesChapter 1 Full Chm556 2018Sabrina100% (1)
- Infra Red SpectroscopiDocument42 pagesInfra Red SpectroscopiMade PrimaNo ratings yet
- Chap 12 - IRDocument30 pagesChap 12 - IRTanvir AhmedNo ratings yet
- Infrared SpectroscopyDocument12 pagesInfrared Spectroscopysanjay sNo ratings yet
- IR Spectroscopy & NMR Spectroscopy MKBDocument44 pagesIR Spectroscopy & NMR Spectroscopy MKBJHidgiwiwNo ratings yet
- Spectros PDFDocument28 pagesSpectros PDFbalajiNo ratings yet
- IR Specroscopy - 2Document16 pagesIR Specroscopy - 2Fatima AhmedNo ratings yet
- Infrared and UVVis SpectrosDocument46 pagesInfrared and UVVis SpectrosOlivia ChoiNo ratings yet
- Chem 84 - IR Spectro Project PaperDocument39 pagesChem 84 - IR Spectro Project PaperCaseyJaneAguilarNo ratings yet
- IR NovDocument53 pagesIR Novmsk3kiidNo ratings yet
- Chap 12ADocument72 pagesChap 12Arareair213No ratings yet
- Infra-Red Spectroscopy: Region Wavelength Range (MM) Wavenumber Range (CM)Document9 pagesInfra-Red Spectroscopy: Region Wavelength Range (MM) Wavenumber Range (CM)Mohit KambojNo ratings yet
- Notes 14C IRDocument6 pagesNotes 14C IRLouie SyNo ratings yet
- Theory of Infra Red AbsorptionDocument108 pagesTheory of Infra Red AbsorptionPreethi IyengarNo ratings yet
- Uv Visible VirenDocument18 pagesUv Visible VirenvirenpatNo ratings yet
- UV - Vis Spectros PDFDocument25 pagesUV - Vis Spectros PDFNelson BarriosNo ratings yet
- NMR Spectroscopy: Koradiya Ketan N. Inorganic Chemistry, Reg. Roll No-5, Bhavnagar University, Bhavnagar-05Document41 pagesNMR Spectroscopy: Koradiya Ketan N. Inorganic Chemistry, Reg. Roll No-5, Bhavnagar University, Bhavnagar-05Koradiya Ketan NNo ratings yet
- Ir 1Document16 pagesIr 1Fatima AhmedNo ratings yet
- 6 10Document5 pages6 10binfa kashafNo ratings yet
- Infrared SpectrosDocument14 pagesInfrared SpectrosV G Viju KumarNo ratings yet
- Spectroscopy - B.Tech First YearDocument26 pagesSpectroscopy - B.Tech First YeartifinNo ratings yet
- Molecular Structure SpectraDocument11 pagesMolecular Structure SpectraAirome CorpuzNo ratings yet
- Introduction To Spectroscopy: I Spectroscopy and The Electromagnetic SpectrumDocument5 pagesIntroduction To Spectroscopy: I Spectroscopy and The Electromagnetic Spectrumbinfa kashafNo ratings yet
- Chapter ThreeDocument32 pagesChapter Threemiesoabdela57No ratings yet
- BY P.Sravanthi M.pharmacy 1styear-Pharmaceutical Analysis TRINITY College of Pharmaceutical SciencesDocument32 pagesBY P.Sravanthi M.pharmacy 1styear-Pharmaceutical Analysis TRINITY College of Pharmaceutical SciencesanithaNo ratings yet
- Infrared Spectroscopy 03Document70 pagesInfrared Spectroscopy 03SowmyaNo ratings yet
- IR SpectrosDocument17 pagesIR SpectrosQasim Jalali NanotiNo ratings yet
- Infrared SpectrosDocument50 pagesInfrared SpectrosmohammedabubakrNo ratings yet
- UV-visible SpectrosDocument61 pagesUV-visible SpectrosMuhammad BilalNo ratings yet
- Electromagnetic RadiationDocument22 pagesElectromagnetic RadiationSelva Kumar0% (1)
- UV VIS Spectroscopy: PHRM 309Document64 pagesUV VIS Spectroscopy: PHRM 309Apurba Sarker Apu100% (1)
- Atomic Structure: Landmarks in The Evolution of Atomic Structure AreDocument11 pagesAtomic Structure: Landmarks in The Evolution of Atomic Structure AreMahmudul IslamNo ratings yet
- Spectroscopy IR NMR Supplemental ReadingDocument14 pagesSpectroscopy IR NMR Supplemental Readingmayagal1707No ratings yet
- IR SpectrosDocument39 pagesIR SpectrosYurîiGislãineNo ratings yet
- 1.UV Spectro - IntroductionDocument49 pages1.UV Spectro - IntroductionRitika GuptaNo ratings yet
- IR SpectrosDocument60 pagesIR SpectrosdeepakNo ratings yet
- Infraredspectroscopy 161018121240Document33 pagesInfraredspectroscopy 161018121240Rajkishor YadavNo ratings yet
- Uv-Visible Spectroscopy: Presented By: Gabriel Engonga and Flora DikeDocument61 pagesUv-Visible Spectroscopy: Presented By: Gabriel Engonga and Flora DikeGabriel EngongaNo ratings yet
- IR Spectroscopy 4Document8 pagesIR Spectroscopy 4alina.tlekkabylova270202No ratings yet
- Topic 7 - Modern Analytical Techniques IDocument2 pagesTopic 7 - Modern Analytical Techniques Ijulian maltoNo ratings yet
- IR and UV SpectroDocument69 pagesIR and UV SpectroSk KumarNo ratings yet
- NMRDocument17 pagesNMRSourabhNo ratings yet
- Lecture Note 16 - Electronic SpectrosDocument24 pagesLecture Note 16 - Electronic SpectrosTbsbi P.No ratings yet
- FTIRDocument79 pagesFTIRshruti shahNo ratings yet
- IR Spectroscopy: Structural Prediction of Organic CompoundsDocument17 pagesIR Spectroscopy: Structural Prediction of Organic CompoundsAdiya Chandak100% (1)
- eBook+Introduction+to+Spectroscopy Pavis 32 121Document7 pageseBook+Introduction+to+Spectroscopy Pavis 32 121devi dwi rustaminingrumNo ratings yet
- Atomic Structure: Chem1101: Chemistry (Eee/Coe)Document11 pagesAtomic Structure: Chem1101: Chemistry (Eee/Coe)Nabil AbdullahNo ratings yet
- Uv Visiblespectroscopyppt 170925144657Document58 pagesUv Visiblespectroscopyppt 170925144657meenu sruthi priyaNo ratings yet
- Electronic SpectrosDocument66 pagesElectronic SpectrosTamilan TamilNo ratings yet
- Infrared SpectrosDocument20 pagesInfrared SpectrosAsim Alaa Al SalehiNo ratings yet
- Chem 442 - Chapter OneDocument26 pagesChem 442 - Chapter OneArindam DasNo ratings yet
- Analisa Fisikokimia Ir FriardiDocument52 pagesAnalisa Fisikokimia Ir FriardiDityaNo ratings yet
- IR and NMR SpectrosDocument13 pagesIR and NMR SpectrosAnand BarapatreNo ratings yet
- Structure, Ligands and Bonding PDFDocument63 pagesStructure, Ligands and Bonding PDFEustance JuanNo ratings yet
- CH 9 Employee and Labour RelationsDocument19 pagesCH 9 Employee and Labour RelationsMark SullivanNo ratings yet
- chm676 Notes PDFDocument43 pageschm676 Notes PDFEustance Juan100% (1)
- Handout1 Vaska CompoundDocument5 pagesHandout1 Vaska CompoundEustance JuanNo ratings yet
- Handout3 Schrock CarbeneDocument3 pagesHandout3 Schrock CarbeneEustance JuanNo ratings yet
- Structure, Ligands and Bonding PDFDocument63 pagesStructure, Ligands and Bonding PDFEustance JuanNo ratings yet
- Outline of Article Analysis LGBTDocument1 pageOutline of Article Analysis LGBTEustance JuanNo ratings yet
- Chap1b MotDocument55 pagesChap1b MotEustance JuanNo ratings yet
- Handout1 Vaska CompoundDocument5 pagesHandout1 Vaska CompoundEustance JuanNo ratings yet
- CHAP1C CO ComplexesDocument39 pagesCHAP1C CO ComplexesEustance JuanNo ratings yet
- Mandarin Script Level 3Document7 pagesMandarin Script Level 3Eustance JuanNo ratings yet
- Top Ten Reason Why Gay/Lesbian Marriage Should Be LegalDocument3 pagesTop Ten Reason Why Gay/Lesbian Marriage Should Be LegalRyanARush100% (1)
- EDITED - VERSION - ANNOTATION - CRITICAL - THINKING - SKILLS - Docx Filename UTF-8''EDITED VERSION ANNOTATION CRITICAL THINKING SKILLSDocument2 pagesEDITED - VERSION - ANNOTATION - CRITICAL - THINKING - SKILLS - Docx Filename UTF-8''EDITED VERSION ANNOTATION CRITICAL THINKING SKILLSEustance JuanNo ratings yet
- Elc550 Assessments Summarised For StudentsDocument2 pagesElc550 Assessments Summarised For StudentsEustance Juan100% (1)
- Chapter 2 CHM510Document48 pagesChapter 2 CHM510Eustance Juan100% (1)
- Chapter 1: An Overview of MarketingDocument17 pagesChapter 1: An Overview of MarketingEustance JuanNo ratings yet
- EDITED - AND - HIGHLIGHTED - Benefits - of - Physical - Play - To - Children - Docx Filename UTF-8''EDITED AND HIGHLIGHTED Benefits of Physical Play To ChildrenDocument1 pageEDITED - AND - HIGHLIGHTED - Benefits - of - Physical - Play - To - Children - Docx Filename UTF-8''EDITED AND HIGHLIGHTED Benefits of Physical Play To ChildrenEustance JuanNo ratings yet
- Lecture PP - Chapter 1.4 - Qualitative AnalysisDocument26 pagesLecture PP - Chapter 1.4 - Qualitative AnalysisEustance JuanNo ratings yet
- Topic9 QualityDocument26 pagesTopic9 QualityEustance JuanNo ratings yet
- Topic10 IslamicManagementDocument16 pagesTopic10 IslamicManagementEustance Juan100% (1)
- 2CHAPTER 2 Aldehydes and Ketones 1Document17 pages2CHAPTER 2 Aldehydes and Ketones 1Eustance JuanNo ratings yet
- Thelook: Feel Good BeautyDocument15 pagesThelook: Feel Good BeautyEmilia RoslanNo ratings yet
- 3CHAPTER 3 Aldehydes and Ketones IIDocument23 pages3CHAPTER 3 Aldehydes and Ketones IIEustance JuanNo ratings yet
- As 2402.1.1-2005 Traction Batteries - Lead-Acid Vented Cells - RequirementsDocument7 pagesAs 2402.1.1-2005 Traction Batteries - Lead-Acid Vented Cells - RequirementsSAI Global - APACNo ratings yet
- Analytical ChemistryDocument6 pagesAnalytical ChemistryRodney Salazar0% (1)
- Arc Geo Mini ManualDocument7 pagesArc Geo Mini ManualhalaNo ratings yet
- ATR Tech WebDocument3 pagesATR Tech WebAudioDuck22No ratings yet
- Chapter05 Systems 2020-03-04Document28 pagesChapter05 Systems 2020-03-04Ali GMNo ratings yet
- Lecture Notes - Number Systems PDFDocument42 pagesLecture Notes - Number Systems PDFjoNo ratings yet
- Automatic Control Systems - Electronics and Communication Engineering Questions and Answers - 1Document9 pagesAutomatic Control Systems - Electronics and Communication Engineering Questions and Answers - 1Rian RiveraNo ratings yet
- DGS-1210-28MP+52MPP E1 Manual v5.00Document127 pagesDGS-1210-28MP+52MPP E1 Manual v5.00Abd AbdNo ratings yet
- Parker Hyd PDFDocument460 pagesParker Hyd PDFAugusto RezendeNo ratings yet
- CMC Database Integrity Declaration (DID) CMCE 1510 03Document1 pageCMC Database Integrity Declaration (DID) CMCE 1510 03Mohd ShahNo ratings yet
- SC-CH4 Manual - Eng - 25 - 09 - 2012Document29 pagesSC-CH4 Manual - Eng - 25 - 09 - 2012Lý Chính ĐạoNo ratings yet
- In Touch With The Medium: Level Monitoring SensorsDocument36 pagesIn Touch With The Medium: Level Monitoring Sensorsm_najmanNo ratings yet
- Wisecube Wis-20: " " " " Precise Shaking IncubatorDocument1 pageWisecube Wis-20: " " " " Precise Shaking IncubatorGrupo de Investigaciones en CatalisisNo ratings yet
- Interpretation of Mass Spectra-4EdDocument329 pagesInterpretation of Mass Spectra-4EdJF Echeverria100% (1)
- Vibration Analysis of Bearings Using FFT Analyzer 59-68Document11 pagesVibration Analysis of Bearings Using FFT Analyzer 59-68Ravi PalaskarNo ratings yet
- General Description Features: Ittybitty RC Timer / OscillatorDocument10 pagesGeneral Description Features: Ittybitty RC Timer / OscillatorIyon ManakarraNo ratings yet
- Data SheetDocument3 pagesData Sheetcontrol9050No ratings yet
- M 312 2000.04 EngelsDocument24 pagesM 312 2000.04 EngelsAlvaro Flores100% (1)
- Manual On Lead Acid BatteriesDocument100 pagesManual On Lead Acid Batterieschandra mouliNo ratings yet
- On GeneratorDocument55 pagesOn GeneratorDileep Reddy91% (11)
- Physics of Ferroelectrics: Pblittlewood January 27, 2002Document26 pagesPhysics of Ferroelectrics: Pblittlewood January 27, 2002Muhammd Usman MalikNo ratings yet
- Terberg Network Structure-3661600586101635Document14 pagesTerberg Network Structure-3661600586101635yousufNo ratings yet
- 02.07 Ecas Can Diagnoza (GB)Document22 pages02.07 Ecas Can Diagnoza (GB)dzadza2No ratings yet
- Delta-2000 Ug PDFDocument138 pagesDelta-2000 Ug PDFJUAN CARLOS BERNAL MEJIANo ratings yet
- 2SC0435T Description and Application ManualDocument16 pages2SC0435T Description and Application ManualRajani Kumar ThirukoluriNo ratings yet
- Laboratory Manual: Analogue and Digital Communication LabDocument18 pagesLaboratory Manual: Analogue and Digital Communication LabZain HaiderNo ratings yet
- Complete EDM Handbook - 10Document8 pagesComplete EDM Handbook - 10peroz_ak47No ratings yet
- Abb Acs800 Crane Control Manualn697 PDFDocument340 pagesAbb Acs800 Crane Control Manualn697 PDFDaniel Tando100% (1)
- Automatic Night Lamp Using LDRDocument3 pagesAutomatic Night Lamp Using LDRMuhammad Haziq100% (2)
- 1 Fundamentals of Piezoelectricity: Lead-Free Piezoelectric MaterialsDocument18 pages1 Fundamentals of Piezoelectricity: Lead-Free Piezoelectric MaterialsJHNo ratings yet