You might also like
- 1.1. An Example of A HMM For Protein Sequences: Output ProbDocument16 pages1.1. An Example of A HMM For Protein Sequences: Output ProbjuanpapoNo ratings yet
- Training ProblemDocument9 pagesTraining ProblemjuanpapoNo ratings yet
- Training ProblemDocument12 pagesTraining ProblemjuanpapoNo ratings yet
- Three Problems of Hidden Markov Models: 1) Scoring ProblemDocument11 pagesThree Problems of Hidden Markov Models: 1) Scoring ProblemjuanpapoNo ratings yet
- Pairwise Sequence AlignmentDocument12 pagesPairwise Sequence Alignmentvanigo1824No ratings yet
- Bio Informatics 005Document4 pagesBio Informatics 005Arooba ShujaNo ratings yet
- Homolgy ModelingDocument19 pagesHomolgy ModelingJainendra JainNo ratings yet
- Homology Modeling, Also Known As Comparative Modeling ofDocument19 pagesHomology Modeling, Also Known As Comparative Modeling ofManoharNo ratings yet
- Protein Structure Similarity: Mlesnick@stanford - EduDocument8 pagesProtein Structure Similarity: Mlesnick@stanford - EdualsreshtyNo ratings yet
- Methods For Applying Multiple Sequence AlignmentDocument17 pagesMethods For Applying Multiple Sequence AlignmentacezeeshanNo ratings yet
- On The Entropy of Protein Families: John P. Barton Arup K. Chakraborty Simona Cocco Hugo Jacquin Rémi MonassonDocument27 pagesOn The Entropy of Protein Families: John P. Barton Arup K. Chakraborty Simona Cocco Hugo Jacquin Rémi MonassonDr. K Lalitha GuruprasadNo ratings yet
- Protein ModellingDocument53 pagesProtein ModellingiluvgermieNo ratings yet
- Multiple Sequence Alignment MSADocument8 pagesMultiple Sequence Alignment MSAAnonymous nUiP53SNo ratings yet
- Comparison of The PAM and BLOSUM Amino Acid Substitution MatricesDocument4 pagesComparison of The PAM and BLOSUM Amino Acid Substitution Matricesmorteza hosseiniNo ratings yet
- Protein Contact Prediction From Amino Acid Co-Evolution Using Convolutional Networks For Graph-Valued ImagesDocument9 pagesProtein Contact Prediction From Amino Acid Co-Evolution Using Convolutional Networks For Graph-Valued ImagesThao LeNo ratings yet
- Summary Bioinformation TechnologyDocument15 pagesSummary Bioinformation TechnologytjNo ratings yet
- Lecture 3 and 4 LSM2241Document6 pagesLecture 3 and 4 LSM2241Koh Yen SinNo ratings yet
- H ' I P P A: MM S Nterpolation of Rotiens For Rofile NalysisDocument10 pagesH ' I P P A: MM S Nterpolation of Rotiens For Rofile NalysisijcseitNo ratings yet
- Bioinformatics IDocument39 pagesBioinformatics IJan Patrick PlatonNo ratings yet
- Bif401 Solved Final Papers 2017Document8 pagesBif401 Solved Final Papers 2017HRrehmanNo ratings yet
- Bookmark This PageDocument35 pagesBookmark This PageAndreea SpiridonNo ratings yet
- Torsion Angles and The Ramachandran PlotDocument2 pagesTorsion Angles and The Ramachandran PlotRitika PaulNo ratings yet
- CME 4425 Introduction To Bioinformatics Algorithms: Zerrin Işık Zerrin@cs - Deu.edu - TRDocument23 pagesCME 4425 Introduction To Bioinformatics Algorithms: Zerrin Işık Zerrin@cs - Deu.edu - TRMahmud KorkmazNo ratings yet
- DH DerivDocument7 pagesDH Derivأحمد دعبسNo ratings yet
- LinearlyDocument12 pagesLinearlywangxiangwen0201No ratings yet
- Chap.3 Protein Structure & Function: TopicsDocument30 pagesChap.3 Protein Structure & Function: TopicssimalihaNo ratings yet
- Local and Global Alignment ResearchpaperDocument12 pagesLocal and Global Alignment ResearchpaperSam GlassmanNo ratings yet
- Polymer StructureDocument4 pagesPolymer StructurePradeep ChaudhariNo ratings yet
- 1 - Sequence Alignment and VisualizationDocument68 pages1 - Sequence Alignment and VisualizationAlisha100% (1)
- Substitution MatrixDocument10 pagesSubstitution MatrixRashmi DhimanNo ratings yet
- Lab Report 1 BME 310Document7 pagesLab Report 1 BME 310Can MunganNo ratings yet
- 322 Refrensi 19Document4 pages322 Refrensi 19bisniskuyNo ratings yet
- Protein3dstructureprediction 161109084221Document30 pagesProtein3dstructureprediction 161109084221Jhanvi SNo ratings yet
- CATH, Bilogical Data Bases, Bioinformatics Data BaseDocument3 pagesCATH, Bilogical Data Bases, Bioinformatics Data BaseRajesh GuruNo ratings yet
- Structure of ProteinDocument72 pagesStructure of ProteinSudipta MandolNo ratings yet
- Iguico, Maria SofiaDocument4 pagesIguico, Maria SofiaDelvin Jan del RosarioNo ratings yet
- Lec06 MPH46 5103Document21 pagesLec06 MPH46 5103Sabith Hasan Khan JoyNo ratings yet
- Bioinformatics Notes - 17Bt54: Module - 4Document48 pagesBioinformatics Notes - 17Bt54: Module - 4Samuel PrinceNo ratings yet
- Alignment CorrectionDocument3 pagesAlignment CorrectionfaisalnadimNo ratings yet
- Using Profile HMM in MSADocument4 pagesUsing Profile HMM in MSAedifoeNo ratings yet
- 7: Source-Based Nomenclature For Copolymers (1985)Document16 pages7: Source-Based Nomenclature For Copolymers (1985)darshanNo ratings yet
- Biochemistry LN05-1Document14 pagesBiochemistry LN05-1Rahaf Al-muhtasebNo ratings yet
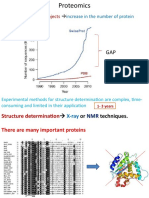
- Genome Sequencing Projects: Increase in The Number of Protein SequencesDocument27 pagesGenome Sequencing Projects: Increase in The Number of Protein SequencesBiju ThomasNo ratings yet
- Mustang PSFB FinalDocument49 pagesMustang PSFB FinalNiranjan BhuvanaratnamNo ratings yet
- On The Accuracy of Homology Modeling and Sequence AlignmentDocument10 pagesOn The Accuracy of Homology Modeling and Sequence AlignmentCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Dot PlotDocument2 pagesDot PlotdarshowlkatNo ratings yet
- L5-Pairwise Seq AlignmentDocument69 pagesL5-Pairwise Seq AlignmentSwayamNo ratings yet
- Amino Acid Substitution Scores: 1 2 N 1 2 N N I 1 I IDocument3 pagesAmino Acid Substitution Scores: 1 2 N 1 2 N N I 1 I ISomnathNo ratings yet
- Protein Structure PredictionDocument52 pagesProtein Structure PredictionMudit MisraNo ratings yet
- Polymer StructureDocument4 pagesPolymer StructureVenkat Reddy YedullaNo ratings yet
- 2013 J Theor Biol 328 77-88Document12 pages2013 J Theor Biol 328 77-88mbrylinskiNo ratings yet
- Protein ModellingDocument15 pagesProtein ModellingNandini KotharkarNo ratings yet
- Statistical Measures To Quantify Similarity Between Molecular Dynamics Simulation TrajectoriesDocument17 pagesStatistical Measures To Quantify Similarity Between Molecular Dynamics Simulation TrajectoriesDiego GranadosNo ratings yet
- Protein Modelling: (Building 3D Models of Proteins)Document19 pagesProtein Modelling: (Building 3D Models of Proteins)rednriNo ratings yet
- Samudrala 1998aDocument22 pagesSamudrala 1998awangxiangwen0201No ratings yet
- Bioinformatics in PAM AND BLOSUMDocument17 pagesBioinformatics in PAM AND BLOSUMgladson100% (15)
- Identification of MOTIFS in Bioactive Peptides PrecursorsDocument14 pagesIdentification of MOTIFS in Bioactive Peptides PrecursorsijbbjournalNo ratings yet
- EOQ ModelsRDocument5 pagesEOQ ModelsRjuanpapoNo ratings yet
- Multiple Sequence AlignmentDocument16 pagesMultiple Sequence AlignmentjuanpapoNo ratings yet
- Future Work.Document20 pagesFuture Work.juanpapoNo ratings yet
- 1.1. Multiple Sequence AlignmentDocument16 pages1.1. Multiple Sequence AlignmentjuanpapoNo ratings yet
- Background: 1.1. DNA - Deoxyribonucleic AcidDocument19 pagesBackground: 1.1. DNA - Deoxyribonucleic AcidjuanpapoNo ratings yet
- Hidden Markov Model (HMM) ArchitectureDocument15 pagesHidden Markov Model (HMM) ArchitecturejuanpapoNo ratings yet
- Primary StructureDocument21 pagesPrimary StructurejuanpapoNo ratings yet
- Limitations of HMMS: P (X P (YDocument1 pageLimitations of HMMS: P (X P (YjuanpapoNo ratings yet
- P (X P (Y: 1. Open Areas For Research in Hmms in BiologyDocument1 pageP (X P (Y: 1. Open Areas For Research in Hmms in BiologyjuanpapoNo ratings yet
- The Central Dogma of Molecular Biology: CctgagccaactattgatgDocument18 pagesThe Central Dogma of Molecular Biology: CctgagccaactattgatgjuanpapoNo ratings yet
- RusobioloDocument21 pagesRusobiolojuanpapoNo ratings yet
- HMM ReportDocument27 pagesHMM ReportŽeljko ŽakulaNo ratings yet
- Biological Background: 1.1. DNA - Deoxyribonucleic AcidDocument19 pagesBiological Background: 1.1. DNA - Deoxyribonucleic AcidjuanpapoNo ratings yet
- Hidden Markov Model (HMM) ArchitectureDocument15 pagesHidden Markov Model (HMM) ArchitecturejuanpapoNo ratings yet
- Biology M PDFDocument13 pagesBiology M PDFjuanpapoNo ratings yet
- Activity Guide and Rubric - Act. 1 Recognition ForumDocument7 pagesActivity Guide and Rubric - Act. 1 Recognition ForumLina Mosquera GuzmanNo ratings yet