You might also like
- Training ProblemDocument9 pagesTraining ProblemjuanpapoNo ratings yet
- Training ProblemDocument12 pagesTraining ProblemjuanpapoNo ratings yet
- Three Problems of Hidden Markov Models: 1) Scoring ProblemDocument11 pagesThree Problems of Hidden Markov Models: 1) Scoring ProblemjuanpapoNo ratings yet
- 1.1. An Example of A HMM For Protein Sequences: Output ProbDocument16 pages1.1. An Example of A HMM For Protein Sequences: Output ProbjuanpapoNo ratings yet
- Using Profile HMM in MSADocument4 pagesUsing Profile HMM in MSAedifoeNo ratings yet
- Homolgy ModelingDocument19 pagesHomolgy ModelingJainendra JainNo ratings yet
- Homology Modeling, Also Known As Comparative Modeling ofDocument19 pagesHomology Modeling, Also Known As Comparative Modeling ofManoharNo ratings yet
- HMMERDocument5 pagesHMMERemma698No ratings yet
- Metamotifs - A Generative Model For Building Families of Nucleotide Position Weight MatricesDocument16 pagesMetamotifs - A Generative Model For Building Families of Nucleotide Position Weight Matricesanon_174376942No ratings yet
- A Mutation Data Matrix For Transmembrane Proteins: LettersDocument7 pagesA Mutation Data Matrix For Transmembrane Proteins: Letterscarlos ArozamenaNo ratings yet
- Re Comb 2003Document16 pagesRe Comb 2003mustafaNo ratings yet
- CG 10 402 PDFDocument14 pagesCG 10 402 PDFAndres GonzalesNo ratings yet
- Protein3dstructureprediction 161109084221Document30 pagesProtein3dstructureprediction 161109084221Jhanvi SNo ratings yet
- Multiple Sequence AlignmentsDocument9 pagesMultiple Sequence Alignmentsvanigo1824No ratings yet
- Protein ModellingDocument53 pagesProtein ModellingiluvgermieNo ratings yet
- 0 AlignMeDocument6 pages0 AlignMeCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Information Sciences: Jun Sun, Xiaojun Wu, Wei Fang, Yangrui Ding, Haixia Long, Webo XuDocument22 pagesInformation Sciences: Jun Sun, Xiaojun Wu, Wei Fang, Yangrui Ding, Haixia Long, Webo XuNeha SharmaNo ratings yet
- J34IJRR ProteinsDocument22 pagesJ34IJRR Proteinscombatps1No ratings yet
- BIF501-Bioinformatics-II Solved Questions FINAL TERM (PAST PAPERS)Document23 pagesBIF501-Bioinformatics-II Solved Questions FINAL TERM (PAST PAPERS)Awais BhuttaNo ratings yet
- Lecture 3 and 4 LSM2241Document6 pagesLecture 3 and 4 LSM2241Koh Yen SinNo ratings yet
- On The Entropy of Protein Families: John P. Barton Arup K. Chakraborty Simona Cocco Hugo Jacquin Rémi MonassonDocument27 pagesOn The Entropy of Protein Families: John P. Barton Arup K. Chakraborty Simona Cocco Hugo Jacquin Rémi MonassonDr. K Lalitha GuruprasadNo ratings yet
- H ' I P P A: MM S Nterpolation of Rotiens For Rofile NalysisDocument10 pagesH ' I P P A: MM S Nterpolation of Rotiens For Rofile NalysisijcseitNo ratings yet
- Local and Global Alignment ResearchpaperDocument12 pagesLocal and Global Alignment ResearchpaperSam GlassmanNo ratings yet
- Theory: Sequence Alignment Is A Process of Aligning Two Sequences To Achieve Maximum Levels ofDocument5 pagesTheory: Sequence Alignment Is A Process of Aligning Two Sequences To Achieve Maximum Levels ofGopi ShankarNo ratings yet
- Micro Array AnalysisDocument29 pagesMicro Array Analysisfelipeg_64No ratings yet
- Sunday, February 8, 2015: Compbio - Biosci.uq - Edu.au/atbDocument2 pagesSunday, February 8, 2015: Compbio - Biosci.uq - Edu.au/atbLalliNo ratings yet
- Pieper NucleicAcidsRes 2004Document6 pagesPieper NucleicAcidsRes 2004ctak2022No ratings yet
- Transmembrane Protein Alignment and Fold Recognition Based On Predicted TopologyDocument9 pagesTransmembrane Protein Alignment and Fold Recognition Based On Predicted TopologyCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Hidden Markov Models Sean R Eddy: Analysis HasDocument5 pagesHidden Markov Models Sean R Eddy: Analysis HasNexus VyasNo ratings yet
- Application of Three Graph Laplacian Based Semi-Supervised Learning Methods To Protein Function Prediction ProblemDocument16 pagesApplication of Three Graph Laplacian Based Semi-Supervised Learning Methods To Protein Function Prediction ProblemijbbjournalNo ratings yet
- Bioinformatics Notes - 17Bt54: Module - 4Document48 pagesBioinformatics Notes - 17Bt54: Module - 4Samuel PrinceNo ratings yet
- Genetic Algorithm HPODocument2 pagesGenetic Algorithm HPOmounaNo ratings yet
- Multiple Sequence Alignment MSADocument8 pagesMultiple Sequence Alignment MSAAnonymous nUiP53SNo ratings yet
- Limitations of HMMS: P (X P (YDocument1 pageLimitations of HMMS: P (X P (YjuanpapoNo ratings yet
- Lec06 MPH46 5103Document21 pagesLec06 MPH46 5103Sabith Hasan Khan JoyNo ratings yet
- A Genetic Algorithm With Clustering For Finding Regulatory Motifs in DNA SequencesDocument5 pagesA Genetic Algorithm With Clustering For Finding Regulatory Motifs in DNA SequencesSoumyabrataBhattacharyya1994No ratings yet
- Bioinformatics AnswersDocument13 pagesBioinformatics AnswersPratibha Patil100% (1)
- Name: Gaurav Bhargava Roll No: IBI2009011Document3 pagesName: Gaurav Bhargava Roll No: IBI2009011Gaurav BhargavaNo ratings yet
- Methods For Applying Multiple Sequence AlignmentDocument17 pagesMethods For Applying Multiple Sequence AlignmentacezeeshanNo ratings yet
- Phylogenetics PDF by Matti Ullah KHan NIaziDocument4 pagesPhylogenetics PDF by Matti Ullah KHan NIazimattiullahkhan860No ratings yet
- Prediction of Loop Geometries Using A Generalized Born Model of Solvation EffectsDocument11 pagesPrediction of Loop Geometries Using A Generalized Born Model of Solvation EffectsLata DeshmukhNo ratings yet
- Simultaneous Topology and Stiffness Identification For Mass-Spring Models Based On FEM Reference DeformationsDocument9 pagesSimultaneous Topology and Stiffness Identification For Mass-Spring Models Based On FEM Reference Deformationsshahuo888No ratings yet
- Identification of Alternative Splice Variants in Aspergillus Flavus Through Comparison of Multiple Tandem Ms Search AlgorithmsDocument10 pagesIdentification of Alternative Splice Variants in Aspergillus Flavus Through Comparison of Multiple Tandem Ms Search AlgorithmsaahhhiiiitttttNo ratings yet
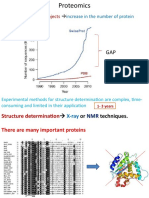
- Genome Sequencing Projects: Increase in The Number of Protein SequencesDocument27 pagesGenome Sequencing Projects: Increase in The Number of Protein SequencesBiju ThomasNo ratings yet
- Multiple Sequence Alignment: Hamid Hamzeiy Izmir Institute of TechnologyDocument6 pagesMultiple Sequence Alignment: Hamid Hamzeiy Izmir Institute of TechnologyHamid HamzeiyNo ratings yet
- Automated Design of The Surface Positions of Protein HelicesDocument9 pagesAutomated Design of The Surface Positions of Protein Helicesaxva1663No ratings yet
- Protein Modelling: (Building 3D Models of Proteins)Document19 pagesProtein Modelling: (Building 3D Models of Proteins)rednriNo ratings yet
- Stam 2004Document12 pagesStam 2004CRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Protein Modeling by Multiple Sequence Threading and Distance GeometryDocument5 pagesProtein Modeling by Multiple Sequence Threading and Distance GeometryLata DeshmukhNo ratings yet
- PSIPREDDocument8 pagesPSIPREDVaibhavi AwaleNo ratings yet
- TMHMMDocument14 pagesTMHMMPubudu GunawardhanaNo ratings yet
- Homology Modeling: Ref: Structural Bioinformatics, P.E Bourne Molecular Modeling, FolkersDocument16 pagesHomology Modeling: Ref: Structural Bioinformatics, P.E Bourne Molecular Modeling, FolkersPallavi VermaNo ratings yet
- Bayesian System IdentificationDocument14 pagesBayesian System IdentificationprawinpsgNo ratings yet
- Comparison of The PAM and BLOSUM Amino Acid Substitution MatricesDocument4 pagesComparison of The PAM and BLOSUM Amino Acid Substitution Matricesmorteza hosseiniNo ratings yet
- MP-T Improving Membrane Protein Alignment For StructureDocument8 pagesMP-T Improving Membrane Protein Alignment For StructureCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Reliability 6Document10 pagesReliability 6Shampa SenNo ratings yet
- The Implementation of A Hidden Markov MoDocument14 pagesThe Implementation of A Hidden Markov MoFito RhNo ratings yet
- 3D Structural Analyses of Bifunctional Protein MdtA For Target Site IdentificationDocument4 pages3D Structural Analyses of Bifunctional Protein MdtA For Target Site IdentificationSabrina JonesNo ratings yet
- Temporal Data Clustering Using Hidden Markov ModelDocument6 pagesTemporal Data Clustering Using Hidden Markov Modelsurendiran123No ratings yet
- Hidden Semi-Markov Models: Theory, Algorithms and ApplicationsFrom EverandHidden Semi-Markov Models: Theory, Algorithms and ApplicationsNo ratings yet
- Shs Stem - Biology 1: Quarter 1 - Module 14Document21 pagesShs Stem - Biology 1: Quarter 1 - Module 14Grace RellosoNo ratings yet
- CELLS - Are the-WPS OfficeDocument2 pagesCELLS - Are the-WPS OfficeAbu DardaNo ratings yet
- CatSper Calcium Channels: 20 Years OnDocument16 pagesCatSper Calcium Channels: 20 Years OnBiblioteca Campus JuriquillaNo ratings yet
- (مكتملة) اسئلة البايو د.ج السمهري (.)Document10 pages(مكتملة) اسئلة البايو د.ج السمهري (.)Ozgan SüleymanNo ratings yet
- Biology 1210 Assigned Study Questions #4: More From Chapter ThreeDocument4 pagesBiology 1210 Assigned Study Questions #4: More From Chapter ThreebigoriginalNo ratings yet
- The Foundations of BiochemistryDocument19 pagesThe Foundations of BiochemistryAsril K100% (1)
- Extraction and Purification Process For Interferon-Beta-1b - Fraunhofer IGBDocument3 pagesExtraction and Purification Process For Interferon-Beta-1b - Fraunhofer IGBJonhy SanchezNo ratings yet
- Cell Surface Markers of T-Cells, B-Cells andDocument6 pagesCell Surface Markers of T-Cells, B-Cells andUdaya KumarNo ratings yet
- Jain Physiology Textbook PDFDocument1,156 pagesJain Physiology Textbook PDFNewgirl596% (28)
- Edexcel Biology Section 1 Powerpoint IgcseDocument21 pagesEdexcel Biology Section 1 Powerpoint IgcseSaba ParkarNo ratings yet
- From Pathogenesis of Acne Vulgaris To Anti-Acne AgentsDocument13 pagesFrom Pathogenesis of Acne Vulgaris To Anti-Acne AgentsNazihan Safitri AlkatiriNo ratings yet
- Negrea Iulia VictorițaDocument9 pagesNegrea Iulia VictorițaMagdalena ApetriiNo ratings yet
- ProteinsDocument2 pagesProteinsSamNo ratings yet
- Advantages and Disadvantages of C4 Plant PhotosynthesisDocument5 pagesAdvantages and Disadvantages of C4 Plant PhotosynthesisAhsan Raza100% (1)
- The Cell QuizDocument7 pagesThe Cell QuizChris GayleNo ratings yet
- The-Cell-Cycle-Worksheet With AnswersDocument5 pagesThe-Cell-Cycle-Worksheet With AnswersNicolai MarquezNo ratings yet
- Fish Immune Responses To Natural Infection With Carp Edema - 2022 - Fish - ShellDocument11 pagesFish Immune Responses To Natural Infection With Carp Edema - 2022 - Fish - Shellfredys seguraNo ratings yet
- Evolution of Cell SignalingDocument3 pagesEvolution of Cell SignalingkatakanNo ratings yet
- Journal Pre-Proofs: Journal of Advanced ResearchDocument55 pagesJournal Pre-Proofs: Journal of Advanced Researchnia rahayu wNo ratings yet
- Molecular Cell Biology Lodish 7th Edition Solutions ManualDocument8 pagesMolecular Cell Biology Lodish 7th Edition Solutions ManualVictoriaWilliamsegtnm100% (83)
- Dna BarcodingDocument17 pagesDna BarcodingJesus Barraza SánchezNo ratings yet
- Error Prone PCRDocument3 pagesError Prone PCRTakwa ShlakiNo ratings yet
- Blakely Ion Channels 2023 Cell and Molec Lecture V2Document29 pagesBlakely Ion Channels 2023 Cell and Molec Lecture V2farzadcop1No ratings yet
- Recombinant DNA MCQDocument6 pagesRecombinant DNA MCQChaze WaldenNo ratings yet
- QP Biology 12 Common Exam Set 3Document8 pagesQP Biology 12 Common Exam Set 3prathishskaNo ratings yet
- Bioinorganic ChemistryDocument11 pagesBioinorganic ChemistryGuru P MNo ratings yet
- Microbiology Principles and Explorations 10th Edition Black Test BankDocument38 pagesMicrobiology Principles and Explorations 10th Edition Black Test Bankcharles90br100% (15)
- RNA Extraction: OverviewDocument3 pagesRNA Extraction: OverviewArturo BasantezNo ratings yet
- Protein Engineering-Methods and ProtocolsDocument347 pagesProtein Engineering-Methods and Protocolslzy100% (1)
- KK5701Document4 pagesKK5701commgmailNo ratings yet