You might also like
- Statistics for Bioinformatics: Methods for Multiple Sequence AlignmentFrom EverandStatistics for Bioinformatics: Methods for Multiple Sequence AlignmentNo ratings yet
- MP-T Improving Membrane Protein Alignment For StructureDocument8 pagesMP-T Improving Membrane Protein Alignment For StructureCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Transmembrane Protein Alignment and Fold Recognition Based On Predicted TopologyDocument9 pagesTransmembrane Protein Alignment and Fold Recognition Based On Predicted TopologyCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Struct GenomDocument7 pagesStruct GenomNaidelyn HernandezNo ratings yet
- On The Accuracy of Homology Modeling and Sequence AlignmentDocument10 pagesOn The Accuracy of Homology Modeling and Sequence AlignmentCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Statistical Measures To Quantify Similarity Between Molecular Dynamics Simulation TrajectoriesDocument17 pagesStatistical Measures To Quantify Similarity Between Molecular Dynamics Simulation TrajectoriesDiego GranadosNo ratings yet
- Structural Category of Protein Membrane Protein: BBL741 Term Paper PresentationDocument7 pagesStructural Category of Protein Membrane Protein: BBL741 Term Paper PresentationMayank SinghNo ratings yet
- Proteins: Remote Homology Detection of Integral Membrane Proteins Using Conserved Sequence FeaturesDocument13 pagesProteins: Remote Homology Detection of Integral Membrane Proteins Using Conserved Sequence FeaturesLavanya Priya SathyanNo ratings yet
- Mustang PSFB FinalDocument49 pagesMustang PSFB FinalNiranjan BhuvanaratnamNo ratings yet
- Graph Based SignatureDocument8 pagesGraph Based SignatureNyasha ChitavatiNo ratings yet
- Muito Detalhada Essa Revisão TranscriptomicaDocument14 pagesMuito Detalhada Essa Revisão TranscriptomicaLeticia PontesNo ratings yet
- 1 s2.0 S0969212611003364 MainDocument11 pages1 s2.0 S0969212611003364 Maintiago almeidaNo ratings yet
- From Genomics To Proteomics: InsightDocument5 pagesFrom Genomics To Proteomics: Insightwadoud aggounNo ratings yet
- Generic CG Folding Aggre ModelsDocument16 pagesGeneric CG Folding Aggre Modelssoumava palitNo ratings yet
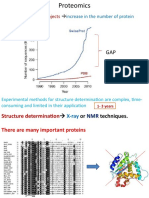
- Genome Sequencing Projects: Increase in The Number of Protein SequencesDocument27 pagesGenome Sequencing Projects: Increase in The Number of Protein SequencesBiju ThomasNo ratings yet
- Integrative Modeling of Membrane-Associated Protein AssembliesDocument12 pagesIntegrative Modeling of Membrane-Associated Protein AssembliesConstanza IsabellaNo ratings yet
- Investigating Transport Proteins by Solid State NMRDocument14 pagesInvestigating Transport Proteins by Solid State NMRAArriiss WizushkiNo ratings yet
- MUMmer PDFDocument8 pagesMUMmer PDFRohith AthreyaNo ratings yet
- Two Sides of A Lipid-Protein StoryDocument13 pagesTwo Sides of A Lipid-Protein StoryFadi Simon de Souza MagalhãesNo ratings yet
- New02 ThelanguageofnatureDocument29 pagesNew02 ThelanguageofnaturesznistvanNo ratings yet
- I-TASSER: A Unified Platform For Automated Protein Structure and Function PredictionDocument14 pagesI-TASSER: A Unified Platform For Automated Protein Structure and Function PredictionDavidNo ratings yet
- Discordancia OMICSDocument7 pagesDiscordancia OMICSLider_rojo88No ratings yet
- Comparison of The PAM and BLOSUM Amino Acid Substitution MatricesDocument4 pagesComparison of The PAM and BLOSUM Amino Acid Substitution Matricesmorteza hosseiniNo ratings yet
- Systems Biology Approaches Applied To Regenerative MedicineDocument9 pagesSystems Biology Approaches Applied To Regenerative MedicineWahyu AndikaNo ratings yet
- A Mutation Data Matrix For Transmembrane Proteins: LettersDocument7 pagesA Mutation Data Matrix For Transmembrane Proteins: Letterscarlos ArozamenaNo ratings yet
- Practical Applications of Proteomics-A Technique For Large-Scale Study of Proteins: An OverviewDocument4 pagesPractical Applications of Proteomics-A Technique For Large-Scale Study of Proteins: An OverviewVishwas gargNo ratings yet
- Post Translational Modifications of Proteins - ReviewDocument20 pagesPost Translational Modifications of Proteins - ReviewNishita GuptaNo ratings yet
- The Ribosome As A Hub For Protein Quality Control - Mol Cell 2013Document11 pagesThe Ribosome As A Hub For Protein Quality Control - Mol Cell 2013aquarius10No ratings yet
- a99Document27 pagesa99Dr. K Lalitha GuruprasadNo ratings yet
- Prognostics of PEM Fuel Cell in A ParticDocument12 pagesPrognostics of PEM Fuel Cell in A ParticJairo Castro FlorianNo ratings yet
- 08-04-16v2 Choosing The Right Protein Biomarker Discovery Tool PDFDocument8 pages08-04-16v2 Choosing The Right Protein Biomarker Discovery Tool PDFCao Minh TríNo ratings yet
- Identification of Functionally Related Enzymes by Learning-to-Rank MethodsDocument13 pagesIdentification of Functionally Related Enzymes by Learning-to-Rank MethodsYahya HashmiNo ratings yet
- Unit 1: Structural GenomicsDocument4 pagesUnit 1: Structural GenomicsLavanya ReddyNo ratings yet
- Protein Unfolding Behavior Studied by Elastic Network ModelDocument11 pagesProtein Unfolding Behavior Studied by Elastic Network ModeljmxnetdjNo ratings yet
- A Model For Competition For Ribosomes in The Cell-M MargaliotDocument29 pagesA Model For Competition For Ribosomes in The Cell-M MargaliotYAAKOV SOLOMONNo ratings yet
- Multi-Scale Modeling of Coarse-Grained Macromolecular LiquidsDocument25 pagesMulti-Scale Modeling of Coarse-Grained Macromolecular LiquidsOsama AdlyNo ratings yet
- Postraductional Prot Review 2021Document20 pagesPostraductional Prot Review 2021Diana CortesNo ratings yet
- Cbar: A Computer Program To Distinguish Plasmid-Derived From Chromosome-Derived Sequence Fragments in Metagenomics DataDocument2 pagesCbar: A Computer Program To Distinguish Plasmid-Derived From Chromosome-Derived Sequence Fragments in Metagenomics Datadumboo21No ratings yet
- Maximum-likelihood tree estimation using codon substitution models with multiple partitionsDocument9 pagesMaximum-likelihood tree estimation using codon substitution models with multiple partitionsPuri RahmaNo ratings yet
- Computational Proteomics: High-Throughput Analysis For Systems BiologyDocument6 pagesComputational Proteomics: High-Throughput Analysis For Systems BiologyMuhammad TahirNo ratings yet
- Bioinformatics 33-22-3549Document9 pagesBioinformatics 33-22-3549donkingdedon30No ratings yet
- Van Deventer Et Al - 2021Document11 pagesVan Deventer Et Al - 2021SusanaNo ratings yet
- Mol - Modelling of BCL-2 FinalDocument37 pagesMol - Modelling of BCL-2 FinalVannakumar SubramanianNo ratings yet
- Cavidad PrediccionDocument9 pagesCavidad PrediccionLuis MartinezNo ratings yet
- Molecular Dynamics Simulations of Membrane Proteins: Philip C. Biggin and Peter J. BondDocument18 pagesMolecular Dynamics Simulations of Membrane Proteins: Philip C. Biggin and Peter J. BondKübra KahveciNo ratings yet
- TMP D73Document15 pagesTMP D73FrontiersNo ratings yet
- Protein To D2O InducesDocument20 pagesProtein To D2O InducesibrahimNo ratings yet
- Genome Organization and Function: A View From Yeast and ArabidopsisDocument13 pagesGenome Organization and Function: A View From Yeast and ArabidopsisJulienne InostrozaNo ratings yet
- EM Structural Survey of Large Protein Complexes in Desulfovibrio VulgarisDocument1 pageEM Structural Survey of Large Protein Complexes in Desulfovibrio Vulgarisapi-77382155No ratings yet
- 0703387Document10 pages0703387dzh12345No ratings yet
- The Role of Protein Structure in Genomics: MinireviewDocument5 pagesThe Role of Protein Structure in Genomics: MinireviewAakriti SharmaNo ratings yet
- Position-Specific Scoring Matrix for Protein Function PredictionDocument8 pagesPosition-Specific Scoring Matrix for Protein Function PredictionW Antonio Muñoz ChNo ratings yet
- Intrinsically Unstructured Proteins: Re-Assessing The Protein Structure-Function ParadigmDocument11 pagesIntrinsically Unstructured Proteins: Re-Assessing The Protein Structure-Function ParadigmJose DanielNo ratings yet
- Generating Conformer Ensembles Using A Multiobjective Genetic AlgorithmDocument13 pagesGenerating Conformer Ensembles Using A Multiobjective Genetic AlgorithmJose GonzalezNo ratings yet
- Reliability 6Document10 pagesReliability 6Shampa SenNo ratings yet
- Flexible MeccanoDocument8 pagesFlexible MeccanoFatima Herranz TrilloNo ratings yet
- A Practical Guide To Molecular Docking and Homology Modelling For Medicinal ChemistsDocument20 pagesA Practical Guide To Molecular Docking and Homology Modelling For Medicinal ChemistsSimona ButanNo ratings yet
- Introduction to Genomics and Omics FieldsDocument16 pagesIntroduction to Genomics and Omics FieldsnavneetNo ratings yet
- Study Protein Interactions Using Chemical Cross-Linkers and Mass SpectrometryDocument13 pagesStudy Protein Interactions Using Chemical Cross-Linkers and Mass SpectrometryJulia MachajNo ratings yet
- Diapositiva 2012tm-Coffee-130709120840-Phpapp02Document24 pagesDiapositiva 2012tm-Coffee-130709120840-Phpapp02CRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- 0 AlignMeDocument6 pages0 AlignMeCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- BALIBASE7REFDocument4 pagesBALIBASE7REFCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Praline 3Document18 pagesPraline 3CRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Survey, Comparison and Evaluation of Cross Platform Mobile Application Development ToolsDocument6 pagesSurvey, Comparison and Evaluation of Cross Platform Mobile Application Development ToolsCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Bioinformatics: PHAT: A Transmembrane-Specific Substitution MatrixDocument7 pagesBioinformatics: PHAT: A Transmembrane-Specific Substitution MatrixCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- PSITM-Coffee A Web SERVERDocument5 pagesPSITM-Coffee A Web SERVERCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Mobile First As A Best Practice in Web Design: Otto VarrelaDocument41 pagesMobile First As A Best Practice in Web Design: Otto VarrelaCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Bioinformatics: PHAT: A Transmembrane-Specific Substitution MatrixDocument7 pagesBioinformatics: PHAT: A Transmembrane-Specific Substitution MatrixCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- SQLLite Database and ListView AdapterDocument19 pagesSQLLite Database and ListView AdapterCRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Lecture 2: Enzymes: Computational Systems BiologyDocument19 pagesLecture 2: Enzymes: Computational Systems Biologyahmad aliNo ratings yet
- Alex Kaivarainen - New Hierarchic Theory of Water and Its Role in Biosystems: The Quantum Psi ProblemDocument54 pagesAlex Kaivarainen - New Hierarchic Theory of Water and Its Role in Biosystems: The Quantum Psi ProblemJoaokaaNo ratings yet
- DNA ShufflingDocument20 pagesDNA ShufflingPreethy ThomasNo ratings yet
- COLLAGENS - Molecular Biology, Diseases, and Potentials For TherapyDocument32 pagesCOLLAGENS - Molecular Biology, Diseases, and Potentials For TherapyRoyaldescentNo ratings yet
- Sewage Pollution and Microbiology PDFDocument311 pagesSewage Pollution and Microbiology PDFjvan migvelNo ratings yet
- Starch and Microbial α-Amylases: From Concepts to Biotechnological ApplicationsDocument30 pagesStarch and Microbial α-Amylases: From Concepts to Biotechnological ApplicationsIndrayana PratamaNo ratings yet
- Protein Structure: Predictive Methods and Experimental MethodologiesDocument33 pagesProtein Structure: Predictive Methods and Experimental MethodologiesmarylaranjoNo ratings yet
- Secondary-Metabolite Biosynthesis and MetabolismDocument372 pagesSecondary-Metabolite Biosynthesis and MetabolismMuhammad IlfanNo ratings yet
- Farmol 2Document27 pagesFarmol 2Ayu DemiNo ratings yet
- The Coronavirus Replicase: Organization, Expression, Processing and FunctionsDocument38 pagesThe Coronavirus Replicase: Organization, Expression, Processing and Functionsfandangos presuntoNo ratings yet
- Munir 2017Document49 pagesMunir 2017Sol AngelNo ratings yet
- Quantitative Models For Microscopic To Macroscopic Biological Macromolecules and TissuesDocument234 pagesQuantitative Models For Microscopic To Macroscopic Biological Macromolecules and TissuesfitoscribdNo ratings yet
- Eabc6405 FullDocument12 pagesEabc6405 Fullvss2012No ratings yet
- Cloning and Expression of Recombinant Sweet Protein Thaumatin II Using Pichia pastoris YeastDocument9 pagesCloning and Expression of Recombinant Sweet Protein Thaumatin II Using Pichia pastoris YeastUdayanidhi RNo ratings yet
- Three-dimensional structure of intensely sweet protein thaumatin revealedDocument4 pagesThree-dimensional structure of intensely sweet protein thaumatin revealedครองศักดา ภัคธนกนกNo ratings yet
- Helicase Enzyme: BiochemistryDocument9 pagesHelicase Enzyme: BiochemistryTayf AlrawINo ratings yet
- UniProt SwissProtDocument4 pagesUniProt SwissProtAriane BeninaNo ratings yet
- A Study on Amylase: A Review of its Structure, Types, Sources and ApplicationsDocument9 pagesA Study on Amylase: A Review of its Structure, Types, Sources and Applicationszaharo putriNo ratings yet
- Ethylene in Plants PDFDocument292 pagesEthylene in Plants PDFGastón Bravo ArrepolNo ratings yet
- CATH, Bilogical Data Bases, Bioinformatics Data BaseDocument3 pagesCATH, Bilogical Data Bases, Bioinformatics Data BaseRajesh GuruNo ratings yet
- Academic Press Encyclopedia of Physical Science and Technology 3rd PolymerDocument339 pagesAcademic Press Encyclopedia of Physical Science and Technology 3rd PolymerBLUECOMANCHE100% (1)
- Rodriguez ViereaDocument51 pagesRodriguez ViereaAwawawawa UwuwuwuwuNo ratings yet
- Docking and Molecular Dynamics Calculations of PyrDocument15 pagesDocking and Molecular Dynamics Calculations of PyrVeliyana Londong AlloNo ratings yet
- Structure and Regulation of Human Phospholipase DDocument10 pagesStructure and Regulation of Human Phospholipase Dxtang2No ratings yet
- Protein Structure Prediction Final ReportDocument27 pagesProtein Structure Prediction Final ReportNithiyaraj VNo ratings yet
- Delta Blast PDFDocument14 pagesDelta Blast PDFHark SranNo ratings yet
- Wikipedia - Readings On Life Evolutionary History V2Document3,688 pagesWikipedia - Readings On Life Evolutionary History V2tariqamin5978No ratings yet
- Protein StructureDocument43 pagesProtein StructureZeyNo ratings yet
- L-Type Calcium Channels: Structure and FunctionsDocument22 pagesL-Type Calcium Channels: Structure and FunctionsrezqNo ratings yet
- ProteinsDocument44 pagesProteinsSusan SylvianingrumNo ratings yet