You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5814)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1092)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (845)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (897)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (540)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (348)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (822)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (401)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Test Bank For Biochemistry 4th Edition Christopher K Mathews Kensal e Van Holde Dean R ApplingDocument12 pagesTest Bank For Biochemistry 4th Edition Christopher K Mathews Kensal e Van Holde Dean R ApplingGeorgeAndersonikwq100% (35)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Che 142 - Final Exam Reviewer 01 Intro Hse in Design: HealthDocument8 pagesChe 142 - Final Exam Reviewer 01 Intro Hse in Design: HealthKaka TsaiNo ratings yet
- PI 100 Report On Cultural ISADocument36 pagesPI 100 Report On Cultural ISAKaka TsaiNo ratings yet
- Notes For NMT & EF ReadingsDocument3 pagesNotes For NMT & EF ReadingsKaka TsaiNo ratings yet
- PI 100 Report On Educational ISADocument37 pagesPI 100 Report On Educational ISAKaka TsaiNo ratings yet
- 2 Vs - 1 Plot: 650 700 F (X) 104.1041490596x - 702.4391748891 R 0.9410704028Document2 pages2 Vs - 1 Plot: 650 700 F (X) 104.1041490596x - 702.4391748891 R 0.9410704028Kaka TsaiNo ratings yet
- GROMACS Installation and Simulation GuideDocument10 pagesGROMACS Installation and Simulation GuideKaka TsaiNo ratings yet
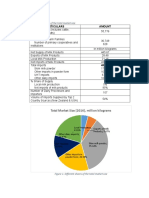
- Total Market Size (2016), Million KilogramsDocument6 pagesTotal Market Size (2016), Million KilogramsKaka TsaiNo ratings yet
- Table 1. Overview of The Total Market Size: Particulars AmountDocument1 pageTable 1. Overview of The Total Market Size: Particulars AmountKaka TsaiNo ratings yet
- Ecology + Bio NotesDocument9 pagesEcology + Bio Notesashra rNo ratings yet
- Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis IIDocument36 pagesSodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis IIYUEALVIN17No ratings yet
- 3 Carbohydrates' StructureDocument33 pages3 Carbohydrates' StructureDilan TeodoroNo ratings yet
- Phospholipid and Sphingosine SynthesisDocument2 pagesPhospholipid and Sphingosine SynthesisAlliah Mae LumanogNo ratings yet
- Topic 3 - Cellular Level of OrganizationDocument6 pagesTopic 3 - Cellular Level of OrganizationAdeyinka OluyoleNo ratings yet
- Understanding Sars-Cov-2-Induced Systemic Amyloidosis: BiorxivDocument4 pagesUnderstanding Sars-Cov-2-Induced Systemic Amyloidosis: BiorxivAntonisNo ratings yet
- Stabilising Forces in Protein Structure: E-Content M.Sc. Zoology (Semester II) CC7-Biochemistry Unit: 3.2Document16 pagesStabilising Forces in Protein Structure: E-Content M.Sc. Zoology (Semester II) CC7-Biochemistry Unit: 3.2paresh kumar sahoo100% (1)
- Enzyme - Wikipedia, The Free EncyclopediaDocument18 pagesEnzyme - Wikipedia, The Free EncyclopediachanackchanackNo ratings yet
- Protein Structure: The Peptide BondDocument13 pagesProtein Structure: The Peptide BondanaNo ratings yet
- Electron Transport ChainDocument35 pagesElectron Transport ChainNusrat JahanNo ratings yet
- DPN - Biochem 1-Exam 2 - 2022Document9 pagesDPN - Biochem 1-Exam 2 - 2022chienyu2002No ratings yet
- 02.06.2018 - 5 - M. Vives Santacana - Principles of PharmacodynamicsDocument46 pages02.06.2018 - 5 - M. Vives Santacana - Principles of PharmacodynamicsPhilippe KinnaerNo ratings yet
- 1.5 Lipids and Protein Exam QuestionsDocument13 pages1.5 Lipids and Protein Exam QuestionsAhmad LuqmanNo ratings yet
- Understanding Omega Fatty AcidsDocument5 pagesUnderstanding Omega Fatty Acidsdrubwang100% (1)
- Alexa Riley - Enzyme Lab ExperimentDocument9 pagesAlexa Riley - Enzyme Lab Experimentapi-553676905No ratings yet
- Aux/IAA Gene Family in Plants: Molecular Structure, Regulation, and FunctionDocument17 pagesAux/IAA Gene Family in Plants: Molecular Structure, Regulation, and FunctionViaNo ratings yet
- 27 - Protein Trafficking - Nuclear TransportDocument12 pages27 - Protein Trafficking - Nuclear TransportHarry DouglasNo ratings yet
- UntitledDocument339 pagesUntitledJOS� FRANCISCO G�MEZ RODR�GUEZNo ratings yet
- J. Gram, J. Jespersen (Auth.), J. Jespersen, R. M. Bertina, F. Haverkate (Eds.) - Laboratory Techniques in Thrombosis - A Manual-Springer Netherlands (1999)Document307 pagesJ. Gram, J. Jespersen (Auth.), J. Jespersen, R. M. Bertina, F. Haverkate (Eds.) - Laboratory Techniques in Thrombosis - A Manual-Springer Netherlands (1999)BipedalJoeNo ratings yet
- Asupan Gizi Dengan Pengendalian Diabetes Pada Diabetisi Tipe Ii Rawat Jalan Di Blu Prof - Dr.R.D.Kandou ManadoDocument11 pagesAsupan Gizi Dengan Pengendalian Diabetes Pada Diabetisi Tipe Ii Rawat Jalan Di Blu Prof - Dr.R.D.Kandou Manadochafeb febiNo ratings yet
- 2nd - Part 3 - Lipid Structure and MetabolismDocument56 pages2nd - Part 3 - Lipid Structure and MetabolismAkbarWirawanNo ratings yet
- PhysioEx Exercise 8 Activity 4Document3 pagesPhysioEx Exercise 8 Activity 4andrea gomez herreraNo ratings yet
- Blood RevisedDocument105 pagesBlood RevisedKanelle SisayanNo ratings yet
- Oxidized LDL (Blog Post)Document10 pagesOxidized LDL (Blog Post)simasNo ratings yet
- Signals and ReceptorsDocument28 pagesSignals and ReceptorsEmilio MurciaNo ratings yet
- Ganga Et Al 2011Document10 pagesGanga Et Al 2011Rachid GangaNo ratings yet
- Sankar Et Al., 2013Document6 pagesSankar Et Al., 2013Natasha MaharaniNo ratings yet
- Protein StructuresDocument6 pagesProtein StructuresKrizzi Dizon GarciaNo ratings yet
- Acute-Phase Protein - WikipediaDocument4 pagesAcute-Phase Protein - WikipediaZACHARIAH MANKIRNo ratings yet