You might also like
- HonorDishonorProcess - Victoria Joy-1 PDFDocument126 pagesHonorDishonorProcess - Victoria Joy-1 PDFarjay1266100% (3)
- RNA-Seq Analysis CourseDocument40 pagesRNA-Seq Analysis Coursejubatus.libroNo ratings yet
- Introduction To Genetic AlgorithmsDocument82 pagesIntroduction To Genetic AlgorithmsMithun Kuniyil100% (6)
- Sand Compaction MethodDocument124 pagesSand Compaction Methodisaych33ze100% (1)
- Amazon Invoice Books 4Document1 pageAmazon Invoice Books 4raghuveer9303No ratings yet
- Why it's important to guard your free timeDocument2 pagesWhy it's important to guard your free timeLaura Camila Garzón Cantor100% (1)
- 2 Integrated MarketingDocument13 pages2 Integrated MarketingPaula Marin CrespoNo ratings yet
- COMP90016 2023 08 Variant Calling IIDocument41 pagesCOMP90016 2023 08 Variant Calling IILynn CHENNo ratings yet
- Molecular Phylogenetic Analysis Methods and ApplicationsDocument35 pagesMolecular Phylogenetic Analysis Methods and ApplicationsDipanjan RayNo ratings yet
- His GaDocument125 pagesHis GaEduard Cosmin UngureanuNo ratings yet
- PhylogeneticsDocument51 pagesPhylogeneticsapi-3807637No ratings yet
- ML Crash CourseDocument10 pagesML Crash Coursepankaj7kalaniaNo ratings yet
- CGE Course JohanneDocument24 pagesCGE Course Johannerazib126No ratings yet
- Sequence Alignment 3Document43 pagesSequence Alignment 3Priyobrota ChyNo ratings yet
- Terminology of Phylogenetic TreesDocument36 pagesTerminology of Phylogenetic TreesDimo PratannaNo ratings yet
- Computational Methods in Phylogenetic Analysis: Tutorial at CSB 2004 Tandy WarnowDocument89 pagesComputational Methods in Phylogenetic Analysis: Tutorial at CSB 2004 Tandy WarnowFemi AlabiNo ratings yet
- Drineas CMU 2012Document59 pagesDrineas CMU 2012juan perez arrikitaunNo ratings yet
- Plenary - WillskyDocument46 pagesPlenary - WillskyRishi RelanNo ratings yet
- Lec8 - Phylogenetic Trees - Till NJDocument36 pagesLec8 - Phylogenetic Trees - Till NJayush sainiNo ratings yet
- QuestionsDocument57 pagesQuestionsbalboinoNo ratings yet
- Slides 9Document62 pagesSlides 9jaa08758No ratings yet
- Cognato q#5Document4 pagesCognato q#5Jesse BarlowNo ratings yet
- Bioinformatics Session16!17!25102021Document39 pagesBioinformatics Session16!17!25102021Rohan RayNo ratings yet
- Soft Computing - Dr. H.S. Hota 28.08.14Document216 pagesSoft Computing - Dr. H.S. Hota 28.08.14Yash 1321No ratings yet
- A Random-Key Genetic Algorithm For The Generalized Traveling Salesman ProblemDocument16 pagesA Random-Key Genetic Algorithm For The Generalized Traveling Salesman ProblemKevin Reyes VegaNo ratings yet
- GNN - PEterDocument96 pagesGNN - PEterzubairNo ratings yet
- Evolutionary Programming Applications To Electrical SystemsDocument15 pagesEvolutionary Programming Applications To Electrical SystemsDeepak SomasundarmamNo ratings yet
- Microarray FullDocument56 pagesMicroarray FullSyed MasudNo ratings yet
- Genetic AlgorithmDocument29 pagesGenetic Algorithm2K20B626 ROHIT KUMAR VERMANo ratings yet
- Lecture 6 Random Forest: From Decision Trees to EnsemblesDocument15 pagesLecture 6 Random Forest: From Decision Trees to EnsemblesLyu PhilipNo ratings yet
- Evolutionary Algorithms: Asst. Prof. Dr. Mohammed Najm AbdullahDocument58 pagesEvolutionary Algorithms: Asst. Prof. Dr. Mohammed Najm AbdullahMuhanad Al-khalisyNo ratings yet
- RNN LSTMDocument72 pagesRNN LSTM5049 Harishchandra KumarNo ratings yet
- Classification Model For Mice Using Genotype Dataset: Assignment-01Document9 pagesClassification Model For Mice Using Genotype Dataset: Assignment-01Dax ValNo ratings yet
- A Two-Dimensional Genetic Algorithm For The Ising Problem: Complex SystDocument7 pagesA Two-Dimensional Genetic Algorithm For The Ising Problem: Complex SystmaucoiNo ratings yet
- Genetic Linkage AnalysisDocument49 pagesGenetic Linkage AnalysisGayatri DaveNo ratings yet
- Probability and Normal DistributionDocument49 pagesProbability and Normal DistributionrolandNo ratings yet
- BTC 506 Phylogenetic AnalysisDocument58 pagesBTC 506 Phylogenetic Analysischijioke NsoforNo ratings yet
- Basic Concepts of Data Mining, Clustering and Genetic AlgorithmsDocument26 pagesBasic Concepts of Data Mining, Clustering and Genetic AlgorithmsharisxyzNo ratings yet
- On the Complexity of SNP Block Partitioning Under the Perfect Phylogeny ModelDocument31 pagesOn the Complexity of SNP Block Partitioning Under the Perfect Phylogeny ModelgrymiNo ratings yet
- Module Q4 2023Document32 pagesModule Q4 2023Stephanie Dawn MagallanesNo ratings yet
- Bioinformatics FinalfinalDocument15 pagesBioinformatics Finalfinalshravani k sNo ratings yet
- Genetic Algorithms: GA Quick OverviewDocument32 pagesGenetic Algorithms: GA Quick OverviewKing RobleNo ratings yet
- Genetic Algorithm IntroductionDocument26 pagesGenetic Algorithm IntroductionDESTELLO GAMINGNo ratings yet
- Using genetic algorithms to find maximal length paths in hypercube graphsDocument7 pagesUsing genetic algorithms to find maximal length paths in hypercube graphsAmandeep KalraNo ratings yet
- Slide 1-14+ Backpropagation (BP) AlgorithmDocument8 pagesSlide 1-14+ Backpropagation (BP) AlgorithmMatt MedrosoNo ratings yet
- L4 - Genetic - AlgorithmDocument138 pagesL4 - Genetic - AlgorithmCHEAT SHEETNo ratings yet
- Computational Genome Analysis: Lecture-4Document60 pagesComputational Genome Analysis: Lecture-4SwayamNo ratings yet
- Random ForestDocument83 pagesRandom ForestBharath Reddy MannemNo ratings yet
- Introduction To Molecular Evolution: Mike Thomas October 3, 2002Document32 pagesIntroduction To Molecular Evolution: Mike Thomas October 3, 2002PINAKIN WNo ratings yet
- Study of Genetic Algorithm An Evolutionary ApproachDocument4 pagesStudy of Genetic Algorithm An Evolutionary ApproachEditor IJRITCCNo ratings yet
- 4701 f17 Final Summary 2Document6 pages4701 f17 Final Summary 2Kaylah KennedyNo ratings yet
- Mathematica Applicanda TemplateDocument8 pagesMathematica Applicanda TemplateFresh Prince Of NigeriaNo ratings yet
- Phylogenetic TreeDocument25 pagesPhylogenetic TreefreelancerhamzaabbasiNo ratings yet
- Gower 1966Document15 pagesGower 1966Ale CLNo ratings yet
- Intro To Phyl o GeneticsDocument44 pagesIntro To Phyl o GeneticsmcinerneyjamesNo ratings yet
- Phylogenetic AnalysisDocument25 pagesPhylogenetic AnalysisRONAK LASHKARINo ratings yet
- Genetic Algorithms Optimize SortingDocument43 pagesGenetic Algorithms Optimize Sortingneena joiceNo ratings yet
- Unit 4Document39 pagesUnit 4namak sung loNo ratings yet
- Jahanshad Solar 2Document51 pagesJahanshad Solar 2api-281558303No ratings yet
- Generalized Linear Mixed Models (Illustrated With R On Bresnan Et Al.'s Datives Data)Document40 pagesGeneralized Linear Mixed Models (Illustrated With R On Bresnan Et Al.'s Datives Data)pauloyyjNo ratings yet
- BDT KSETA FreudenstadtDocument32 pagesBDT KSETA FreudenstadtPicasbrancasNo ratings yet
- Stats and Data AnalysisDocument49 pagesStats and Data AnalysisEman YahiaNo ratings yet
- B. Tech, Batch, 2 Semester Subject Code: 18CS3064: Subject: Big Data OptimizationDocument11 pagesB. Tech, Batch, 2 Semester Subject Code: 18CS3064: Subject: Big Data Optimizationkrishnasai tadiboinaNo ratings yet
- A Sampling of Various Other Learning MethodsDocument34 pagesA Sampling of Various Other Learning MethodsDhriti TuliNo ratings yet
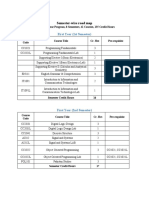
- Semester-Wise Road Map: First Year (1st Semester)Document4 pagesSemester-Wise Road Map: First Year (1st Semester)Hashim AliNo ratings yet
- SpecialGCMeetingData October 2020Document3 pagesSpecialGCMeetingData October 2020Hashim AliNo ratings yet
- Some TextDocument1 pageSome TextHashim AliNo ratings yet
- Original Image Filtered Image Original-Filtered ImageDocument1 pageOriginal Image Filtered Image Original-Filtered ImageHashim AliNo ratings yet
- CS101L Manual V1.1Document184 pagesCS101L Manual V1.1Muaz ShafiqNo ratings yet
- Semester-Wise Road Map: First Year (1st Semester)Document4 pagesSemester-Wise Road Map: First Year (1st Semester)Hashim AliNo ratings yet
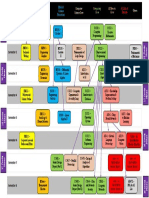
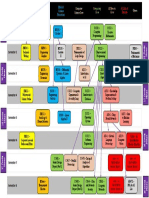
- Image 1 - BS in AI Dependency Graph PDFDocument1 pageImage 1 - BS in AI Dependency Graph PDFHashim Ali100% (1)
- CommentsDocument1 pageCommentsHashim AliNo ratings yet
- Image 1 - BS in AI Dependency GraphDocument1 pageImage 1 - BS in AI Dependency GraphHashim AliNo ratings yet
- Fcse 1Document22 pagesFcse 1Hashim AliNo ratings yet
- Course Descriptions Core Courses: Undergraduate Prospectus 2020Document15 pagesCourse Descriptions Core Courses: Undergraduate Prospectus 2020Hashim AliNo ratings yet
- Spring 2019 Course Checklist CSDocument3 pagesSpring 2019 Course Checklist CSHashim AliNo ratings yet
- Program Educational Objectives (Peos) of Bs (Ai)Document7 pagesProgram Educational Objectives (Peos) of Bs (Ai)Hashim AliNo ratings yet
- Artificial Intelligence - Semester-Wise BreakdownDocument6 pagesArtificial Intelligence - Semester-Wise BreakdownHashim AliNo ratings yet
- Prospectus 2015Document164 pagesProspectus 2015malik karim dadNo ratings yet
- CH 4Document40 pagesCH 4KarimNo ratings yet
- Dreams of CalculusDocument172 pagesDreams of CalculusHashim AliNo ratings yet
- Desk PiDocument21 pagesDesk PiThan LwinNo ratings yet
- Q3 Curriculum Map - Mathematics 10Document3 pagesQ3 Curriculum Map - Mathematics 10Manilyn BaltazarNo ratings yet
- CS6711 Security Lab ManualDocument84 pagesCS6711 Security Lab ManualGanesh KumarNo ratings yet
- S 1804 2019 (E) - 0Document9 pagesS 1804 2019 (E) - 0Juan Agustin CuadraNo ratings yet
- Lecture01 PushkarDocument27 pagesLecture01 PushkarabcdNo ratings yet
- Top-Down DesignDocument18 pagesTop-Down DesignNguyễn Duy ThôngNo ratings yet
- Compare The Political System of Kazakhstan, USA, UK PresentationDocument19 pagesCompare The Political System of Kazakhstan, USA, UK PresentationAiganym OmiraliNo ratings yet
- Parts of The Analog MultitesterDocument4 pagesParts of The Analog MultitesterDestiny Marasigan CanacanNo ratings yet
- Engr2227 Apr03Document10 pagesEngr2227 Apr03Mohamed AlqaisiNo ratings yet
- Chinese in The PHDocument15 pagesChinese in The PHMandalihan GepersonNo ratings yet
- Classification of AnimalsDocument6 pagesClassification of Animalsapi-282695651No ratings yet
- Cubic Spline Tutorial v3Document6 pagesCubic Spline Tutorial v3Praveen SrivastavaNo ratings yet
- Cebuano LanguageDocument15 pagesCebuano LanguageIsla PageNo ratings yet
- Ventilator Modes - WEANINGDocument3 pagesVentilator Modes - WEANINGAlaa OmarNo ratings yet
- Radio Codes and ConventionsDocument2 pagesRadio Codes and Conventionsapi-570661298No ratings yet
- Joe Ann MarcellanaDocument17 pagesJoe Ann MarcellanarudyNo ratings yet
- Is Iso 2692-1992Document24 pagesIs Iso 2692-1992mwasicNo ratings yet
- St. Anthony College Calapan City Syllabus: Course DescriptionDocument6 pagesSt. Anthony College Calapan City Syllabus: Course DescriptionAce HorladorNo ratings yet
- A Review of The Management of Cerebral Vasospasm After Aneurysmal Subarachnoid HemorrhageDocument15 pagesA Review of The Management of Cerebral Vasospasm After Aneurysmal Subarachnoid HemorrhageAlanNo ratings yet
- 2020.07.31 Marchese Declaration With ExhibitsDocument103 pages2020.07.31 Marchese Declaration With Exhibitsheather valenzuelaNo ratings yet
- Basketball 2011: Johnson CountyDocument25 pagesBasketball 2011: Johnson CountyctrnewsNo ratings yet
- Atomic Structure QuestionsDocument1 pageAtomic Structure QuestionsJames MungallNo ratings yet
- WISECO 2011 Complete CatalogDocument131 pagesWISECO 2011 Complete CatalogfishuenntNo ratings yet
- Cisco Series SWCFG Xe 16 12 XDocument416 pagesCisco Series SWCFG Xe 16 12 XWagner SantiagoNo ratings yet
- If Sentences Type 1 First Type Conditionals Grammar Drills - 119169Document2 pagesIf Sentences Type 1 First Type Conditionals Grammar Drills - 119169Ivanciu DanNo ratings yet