You might also like
- MECEE BL Modal Entrance Exam Solutions (Set-XV B 2079-04-14Document12 pagesMECEE BL Modal Entrance Exam Solutions (Set-XV B 2079-04-14Sameer KhanNo ratings yet
- D0685 Biology Paper2Document9 pagesD0685 Biology Paper2Aryan SinghNo ratings yet
- Cell Culture1Document2 pagesCell Culture1Marivic Parrocho FrancoNo ratings yet
- Rat Adult Stem Cells (Marrow Stromal Cells) Engraft and Differentiate in Chick Embryos Without Evidence of Cell FusionDocument4 pagesRat Adult Stem Cells (Marrow Stromal Cells) Engraft and Differentiate in Chick Embryos Without Evidence of Cell Fusion1alfredo12100% (1)
- Blood NotesDocument19 pagesBlood NotesLekhana BankiNo ratings yet
- In Vitro Cultivation of Fasciola He'Patica Metacercariae and of Partially Developed Flukes Recovered From MiceDocument7 pagesIn Vitro Cultivation of Fasciola He'Patica Metacercariae and of Partially Developed Flukes Recovered From MiceGuisela Huamanta AguilarNo ratings yet
- The Veterinary Journal: V. Hall, K. Hinrichs, G. Lazzari, D.H. Betts, P. HyttelDocument15 pagesThe Veterinary Journal: V. Hall, K. Hinrichs, G. Lazzari, D.H. Betts, P. HyttelJuniClaudia13No ratings yet
- Induced Pluripotency and Epigenetic Reprogramming: Konrad Hochedlinger and Rudolf JaenischDocument26 pagesInduced Pluripotency and Epigenetic Reprogramming: Konrad Hochedlinger and Rudolf JaenischnembutalNo ratings yet
- JurnalDocument13 pagesJurnalparkc6922No ratings yet
- Ehics and Biomedical SkillsDocument118 pagesEhics and Biomedical SkillsBiology BảoNo ratings yet
- generation iPSC from BJ cells JOVE protocolDocument3 pagesgeneration iPSC from BJ cells JOVE protocolrajeshkundapur123No ratings yet
- 2-SC-therapy and iPS cellsDocument46 pages2-SC-therapy and iPS cellsmarinaNo ratings yet
- Hematology ReviewerDocument4 pagesHematology ReviewerAbigail Puno100% (1)
- Stem Cells: Dr. Valerie Bracchi-RicardDocument49 pagesStem Cells: Dr. Valerie Bracchi-RicardE12H12No ratings yet
- CAP 4 Development of The MammalianDocument12 pagesCAP 4 Development of The Mammaliangilson_pessoaNo ratings yet
- Chatterjee 2015Document17 pagesChatterjee 2015SREEHARI SREEKUMAR 19BBT0117No ratings yet
- Proteomics of Reproductive Fluids and Sperm Cells of Rams - 1-s2.0-S0093691X20305355-mainDocument10 pagesProteomics of Reproductive Fluids and Sperm Cells of Rams - 1-s2.0-S0093691X20305355-mainNadia MirandaNo ratings yet
- Animal BiotechnologyDocument31 pagesAnimal BiotechnologyaparnayadavNo ratings yet
- Stem Cells - overview: Embryonic stem cell therapy for spinal cord injury faces delaysDocument39 pagesStem Cells - overview: Embryonic stem cell therapy for spinal cord injury faces delaysubaidu dikkoNo ratings yet
- Common Model Exam Set-IX (2078-7-6) SolDocument12 pagesCommon Model Exam Set-IX (2078-7-6) SolPrepladder ChyNo ratings yet
- @artigo Novo 2Document6 pages@artigo Novo 2Beatrice MacenteNo ratings yet
- Development of The Avian Immune System: October 2013Document20 pagesDevelopment of The Avian Immune System: October 2013Ferdinand Prayogo Cahyo SantosoNo ratings yet
- Solved QuestionsDocument30 pagesSolved QuestionsTeiborlin MarngarNo ratings yet
- Common Model Exam Set-XIII - B - 2079-3-32 - SolutionDocument12 pagesCommon Model Exam Set-XIII - B - 2079-3-32 - SolutionSameer KhanNo ratings yet
- Evaluation Component: Mid Semester (CB)Document5 pagesEvaluation Component: Mid Semester (CB)Aaryan GuptaNo ratings yet
- Growth of Fish Cell Lines on Microcarriers and Production of Channel Catfish VirusDocument14 pagesGrowth of Fish Cell Lines on Microcarriers and Production of Channel Catfish VirusJonathan RedricoNo ratings yet
- +2 Zoology WorksheetsDocument33 pages+2 Zoology WorksheetsLena AlbertNo ratings yet
- Cbse SR Jee Apex & Neet Wisdom Preboard Biology KeyDocument5 pagesCbse SR Jee Apex & Neet Wisdom Preboard Biology KeyAshwina JaikrishnanNo ratings yet
- What happens during fertilisationDocument20 pagesWhat happens during fertilisationelizabeth_rk_wattNo ratings yet
- Transgenic Organisms2017Document36 pagesTransgenic Organisms2017Anastacio Elston EusoresNo ratings yet
- 1r41ai10801601 Fong Part16Document3 pages1r41ai10801601 Fong Part16apple12zsxxNo ratings yet
- Developmental Biology Test QuestionsDocument73 pagesDevelopmental Biology Test QuestionsPearl PascuaNo ratings yet
- 5665 FullDocument4 pages5665 FullFé Rî ÉlNo ratings yet
- الاسئلة بعد التعديلDocument3 pagesالاسئلة بعد التعديلZainab HasanNo ratings yet
- DR Ajay Popli: Consultant:-Minimally Invasive Spine & Orthopedics SurgeryDocument22 pagesDR Ajay Popli: Consultant:-Minimally Invasive Spine & Orthopedics SurgeryAjay PopliNo ratings yet
- Cloning Human BeingsDocument310 pagesCloning Human BeingsThe Hastings CenterNo ratings yet
- HGL2010Document9 pagesHGL2010api-28650133No ratings yet
- Transgenic Animal Bioreactors in Biotechnology of Blood Proteins and ProductionDocument54 pagesTransgenic Animal Bioreactors in Biotechnology of Blood Proteins and ProductionZeeshan AnjumNo ratings yet
- Production of Transgenic Goats Expressing Human Coagulation Factor IX in The Mammary Glands After Nuclear Transfer Using Transfected Fetal Fibroblast CellsDocument9 pagesProduction of Transgenic Goats Expressing Human Coagulation Factor IX in The Mammary Glands After Nuclear Transfer Using Transfected Fetal Fibroblast CellsMaira QuintanaNo ratings yet
- 1 s2.0 S0092867418300576 MainextDocument17 pages1 s2.0 S0092867418300576 MainextJOHN ALEJANDRO DIAZ RODRIGUEZNo ratings yet
- Developmental Control of Macrophage FunctionDocument11 pagesDevelopmental Control of Macrophage FunctionMaria SunflowerNo ratings yet
- 259 Full PDFDocument12 pages259 Full PDFNehaNo ratings yet
- RST B 20090196Document15 pagesRST B 20090196baheNo ratings yet
- PhysiologicalDocument5 pagesPhysiologicalLalmohan KarNo ratings yet
- Match Cell Organelles and Their FunctionsDocument68 pagesMatch Cell Organelles and Their FunctionsViswaNo ratings yet
- 2nd Pu Mcqs For Preparatory ExamDocument9 pages2nd Pu Mcqs For Preparatory ExamMaster manNo ratings yet
- The Mouse Epididymal Transcriptome: Transcriptional Profiling of Segmental Gene Expression in The EpididymisDocument10 pagesThe Mouse Epididymal Transcriptome: Transcriptional Profiling of Segmental Gene Expression in The EpididymisAhmad SolihinNo ratings yet
- Darwinian Natural Selection - Lecture 3Document43 pagesDarwinian Natural Selection - Lecture 3Brian PaguiaNo ratings yet
- Assignment Class-12 Subject:Bio - Zoology Unit 1: Chapter 2. Human ReproductionDocument6 pagesAssignment Class-12 Subject:Bio - Zoology Unit 1: Chapter 2. Human ReproductionjohnsonNo ratings yet
- iPSC Guide by ATCC PDFDocument39 pagesiPSC Guide by ATCC PDFvadhaNo ratings yet
- Gomez 2004 Birth of African Wildcat Cloned Kittens Born From Domestic Cats1Document14 pagesGomez 2004 Birth of African Wildcat Cloned Kittens Born From Domestic Cats1Revio Reidi PutraNo ratings yet
- Stem Cell Technology: November 2017Document43 pagesStem Cell Technology: November 2017priyanka boopathyNo ratings yet
- Mechanisms of Morphogenesis and Pattern FormationDocument37 pagesMechanisms of Morphogenesis and Pattern Formationstoic_2010No ratings yet
- COMED K 2016 - Normal QuestionDocument72 pagesCOMED K 2016 - Normal QuestionGowri ShankarNo ratings yet
- 8 ScienceDocument13 pages8 SciencesheljyalexNo ratings yet
- History of Animal Cell CultureDocument30 pagesHistory of Animal Cell CultureCatleah ZamoraNo ratings yet
- Biology One Mark QuestionDocument12 pagesBiology One Mark Questionvishali GNo ratings yet
- Rohwerder 2003Document10 pagesRohwerder 2003Dwi Yerlis RahmiNo ratings yet
- Marchenko 2016Document5 pagesMarchenko 2016Dwi Yerlis RahmiNo ratings yet
- BG 8 B Inggris AyomadrasahDocument2 pagesBG 8 B Inggris AyomadrasahDwi Yerlis RahmiNo ratings yet
- 10 1016@j Eti 2019 100369 PDFDocument39 pages10 1016@j Eti 2019 100369 PDFDwi Yerlis RahmiNo ratings yet
- 6324 12103 1 SM PDFDocument6 pages6324 12103 1 SM PDFDwi Yerlis RahmiNo ratings yet
- 10 1016@j Eti 2019 100369 PDFDocument39 pages10 1016@j Eti 2019 100369 PDFDwi Yerlis RahmiNo ratings yet
- Dwi Yerlis Rahmi - Biodiesel Minyak Biji KaretDocument21 pagesDwi Yerlis Rahmi - Biodiesel Minyak Biji KaretDwi Yerlis RahmiNo ratings yet
- Daftar Nama Siswa Kelas Viii TP-LKDocument2 pagesDaftar Nama Siswa Kelas Viii TP-LKDwi Yerlis RahmiNo ratings yet
- Bioceramics Synthetic OptionsDocument2 pagesBioceramics Synthetic OptionsDwi Yerlis RahmiNo ratings yet
- Chapter IIDocument10 pagesChapter IIDwi Yerlis RahmiNo ratings yet
- Wide LL 1984Document9 pagesWide LL 1984Dwi Yerlis RahmiNo ratings yet
- Fee2014 PDFDocument41 pagesFee2014 PDFKingSandwichNo ratings yet
- Yu 2008Document7 pagesYu 2008Dwi Yerlis RahmiNo ratings yet
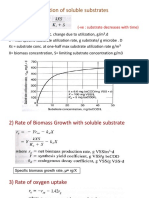
- 1) Rate of Utilization of Soluble SubstratesDocument5 pages1) Rate of Utilization of Soluble SubstratesDwi Yerlis RahmiNo ratings yet
- Chapter 4Document10 pagesChapter 4Dwi Yerlis RahmiNo ratings yet
- Nurul - Afifah (Bio1)Document6 pagesNurul - Afifah (Bio1)Dwi Yerlis RahmiNo ratings yet
- Absorption & Stripping Design OptimizationDocument11 pagesAbsorption & Stripping Design OptimizationWaleed AkbarNo ratings yet
- SDS 20 Pages - 153Document30 pagesSDS 20 Pages - 153Dwi Yerlis RahmiNo ratings yet
- STDDocument2 pagesSTDDwi Yerlis RahmiNo ratings yet
- Biotechnology On The Treatment of WastesDocument10 pagesBiotechnology On The Treatment of WastesKevin CooperNo ratings yet
- Article1380817563 - Buyukgungor and GurelDocument7 pagesArticle1380817563 - Buyukgungor and GurelDwi Yerlis RahmiNo ratings yet
- Chapter 12 - Surface Water TreatmentDocument36 pagesChapter 12 - Surface Water TreatmentDwi Yerlis RahmiNo ratings yet
- Benzene Specification: Properties Test Method Unit SpecificationsDocument1 pageBenzene Specification: Properties Test Method Unit SpecificationsDwi Yerlis RahmiNo ratings yet
- Handbook For Cell Culture Basics (Gibco)Document62 pagesHandbook For Cell Culture Basics (Gibco)Cesar PuentesNo ratings yet
- Creep DescriptionDocument11 pagesCreep DescriptionGerry AnandaNo ratings yet
- Lampiran A BaganDocument2 pagesLampiran A BaganDwi Yerlis RahmiNo ratings yet
- MF7134 MSDSDocument5 pagesMF7134 MSDSDwi Yerlis RahmiNo ratings yet
- MF7134 PSDocument4 pagesMF7134 PSDwi Yerlis RahmiNo ratings yet
- Aerogel Synthesis and ApplicationDocument55 pagesAerogel Synthesis and ApplicationgonzalezpuertoNo ratings yet
- In This Phase, 2 ATP Are Used.: 1 Glucose Is Converted Into 2 Glyceraldehyde-3-PhosphateDocument8 pagesIn This Phase, 2 ATP Are Used.: 1 Glucose Is Converted Into 2 Glyceraldehyde-3-PhosphateGia HoàngNo ratings yet
- Food Biochemistry and Food ProcessingDocument761 pagesFood Biochemistry and Food ProcessingOleg100% (14)
- BMSC 230 - Modules 1 & 2 Lecture NotesDocument21 pagesBMSC 230 - Modules 1 & 2 Lecture NotescamrynNo ratings yet
- PHD Thesis The Production of Bacteriocins From Lactic Acid Bacteria 2011Document2 pagesPHD Thesis The Production of Bacteriocins From Lactic Acid Bacteria 2011Myrto-Panagiota ZacharofNo ratings yet
- Fermentación BatchDocument8 pagesFermentación BatchJennifer A. PatiñoNo ratings yet
- 1 s2.0 S0362028X23014928 MainDocument9 pages1 s2.0 S0362028X23014928 MainElizabeth BennetNo ratings yet
- ChucrutDocument6 pagesChucrutPAOLANo ratings yet
- Chapter-IV Fermentation PDFDocument8 pagesChapter-IV Fermentation PDFJignesh SolankiNo ratings yet
- QUESTION Bank Biochemical EngineeringDocument4 pagesQUESTION Bank Biochemical EngineeringMilito GilbertoNo ratings yet
- Cyclist Australia - Issue 7Document132 pagesCyclist Australia - Issue 7pmathiasNo ratings yet
- Goljan Audio Transcripts (125p)Document125 pagesGoljan Audio Transcripts (125p)danielastep1No ratings yet
- The Genera of Lactic Acid Bacteria - Wood (1995)Document414 pagesThe Genera of Lactic Acid Bacteria - Wood (1995)ISRAELNo ratings yet
- Calculation of Substrate Oxidation Rates in Vivo From Gaseous ExchangeDocument7 pagesCalculation of Substrate Oxidation Rates in Vivo From Gaseous ExchangeRodrigo CastilloNo ratings yet
- Lesson Plan in Jan. 13 Mole Concept Grade 9Document5 pagesLesson Plan in Jan. 13 Mole Concept Grade 9Edessa MasinasNo ratings yet
- Blood Lactate Response To Overtraining in Male Endurance AthletesDocument8 pagesBlood Lactate Response To Overtraining in Male Endurance AthletesGerasimos29No ratings yet
- Modeling of Batch Fermentation Kinetics For Succinic Acid Production by Mannheneimia SucciniciproducensDocument9 pagesModeling of Batch Fermentation Kinetics For Succinic Acid Production by Mannheneimia SucciniciproducensRyuuara Az-ZahraNo ratings yet
- Causes HyperlactemiaDocument5 pagesCauses HyperlactemiaArvin ReinaldoNo ratings yet
- Fermentation PathwaysDocument28 pagesFermentation PathwaysNesma NsrNo ratings yet
- Waterlogging - WPS OfficeDocument5 pagesWaterlogging - WPS OfficeReenam MatloobNo ratings yet
- Milk (Contamination and Sources)Document6 pagesMilk (Contamination and Sources)GURJEET SINGHNo ratings yet
- Esayas AssefaDocument80 pagesEsayas AssefahabtastaNo ratings yet
- Concept Importance and Types of Starter Cultures in Dairy IndustryDocument21 pagesConcept Importance and Types of Starter Cultures in Dairy IndustryRohit PandeyNo ratings yet
- Shrimp ShellDocument11 pagesShrimp ShellSaranya RathanNo ratings yet
- Understanding Lactate in the EDDocument7 pagesUnderstanding Lactate in the EDSyed Shahrul Naz SyedNo ratings yet
- Recovery and Purification of Lactic Acid From Fermentation BrothDocument185 pagesRecovery and Purification of Lactic Acid From Fermentation BrothBilli CostanNo ratings yet
- JJJ ThursdayDocument47 pagesJJJ ThursdayhozhabrNo ratings yet
- Worksheet FermentationDocument5 pagesWorksheet FermentationRyan De AlloNo ratings yet
- United States Patent (10) Patent No.: US 6,326,458 B1Document26 pagesUnited States Patent (10) Patent No.: US 6,326,458 B1Citra Adelina SitorusNo ratings yet
- Bifidobacterium Spp. and Lactobacillus Acidophilus - Biological, Biochemical, Technological and Therapeutical - REVIEW PDFDocument19 pagesBifidobacterium Spp. and Lactobacillus Acidophilus - Biological, Biochemical, Technological and Therapeutical - REVIEW PDFmilu1312No ratings yet