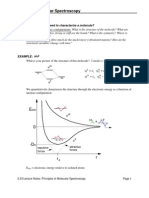
You might also like
- Neutron Proton ScatteringDocument7 pagesNeutron Proton ScatteringDebayan DasguptaNo ratings yet
- Conversion KVA KWDocument1 pageConversion KVA KWAlfonso Giménez100% (4)
- Silicon - Controlled Rectifiers: Group MembersDocument13 pagesSilicon - Controlled Rectifiers: Group MembersMartin John Ramirez100% (1)
- MolecularEnergyLevelsNotes PDFDocument5 pagesMolecularEnergyLevelsNotes PDFLorena Monterrosas PérezNo ratings yet
- Molecular Energy Levels NotesDocument5 pagesMolecular Energy Levels Notesskrim2No ratings yet
- Microwave Spectroscopy-1Document35 pagesMicrowave Spectroscopy-1surajjamal111No ratings yet
- Rotational-Vibrational Spectra The Oscillating RotatorDocument5 pagesRotational-Vibrational Spectra The Oscillating RotatortamilselviNo ratings yet
- Rotational-Vibrational SpectrosDocument13 pagesRotational-Vibrational Spectrosmarosh.marwa.slivaniNo ratings yet
- Rovibrational SpectrosDocument9 pagesRovibrational SpectrosPrasad DurgaNo ratings yet
- Molecular Spectroscopy Crib Sheet GuideDocument4 pagesMolecular Spectroscopy Crib Sheet GuidedjevNo ratings yet
- Chemistry 1A Spectroscopy.: Prof. Mike Ashfold (S305) (Mike - Ashfold@bris - Ac.uk)Document63 pagesChemistry 1A Spectroscopy.: Prof. Mike Ashfold (S305) (Mike - Ashfold@bris - Ac.uk)Abd El-Fattah Mohamed OufNo ratings yet
- Lecture 3Document18 pagesLecture 3Balaraju BanothNo ratings yet
- Type of SpectrumDocument5 pagesType of Spectrumfiza choudharyNo ratings yet
- 3 Assumptions and ApproximationsDocument5 pages3 Assumptions and ApproximationsJack RyderNo ratings yet
- Spectroscopy Rotational SpectrosDocument7 pagesSpectroscopy Rotational SpectrosMONERAH D ALOSAIMINo ratings yet
- Electronic Absorption Spectroscopy: M, E, A ADocument8 pagesElectronic Absorption Spectroscopy: M, E, A ANiko DidicNo ratings yet
- Maxwell's Equations and the Transverse Nature of Light WavesDocument24 pagesMaxwell's Equations and the Transverse Nature of Light WavesFarizalNo ratings yet
- Principles of Molecular SpectrosDocument12 pagesPrinciples of Molecular SpectrosShaheentariq_4No ratings yet
- Wave EquationDocument14 pagesWave EquationbigouzypolNo ratings yet
- Spectroscopy I (1a)Document6 pagesSpectroscopy I (1a)shaharhr1No ratings yet
- Vibrasi Dari AtkinsDocument7 pagesVibrasi Dari AtkinsFawzia AuliaNo ratings yet
- Exp4 IodineDocument6 pagesExp4 IodinePraveen AKNo ratings yet
- P2214 Homework 14 Solutions - Spring 2011Document7 pagesP2214 Homework 14 Solutions - Spring 2011calcyeeNo ratings yet
- APhO2001 Theory Prob 2 PDFDocument2 pagesAPhO2001 Theory Prob 2 PDFRanojit barmanNo ratings yet
- 604 BookDocument82 pages604 BooknewzadNo ratings yet
- B ex x A B e (A η B ex x A A B: i j p i j P P P i jDocument1 pageB ex x A B e (A η B ex x A A B: i j p i j P P P i jNeoHoodaNo ratings yet
- Spectroscopy - Rotational Spectroscopy - WikiversityDocument9 pagesSpectroscopy - Rotational Spectroscopy - WikiversityDr. Gazi Jahirul ISlamNo ratings yet
- Solid State SatyabhamaDocument106 pagesSolid State Satyabhamawname384No ratings yet
- 2.57 Nano-to-Macro Transport Processes Fall 2004: E HN MDocument10 pages2.57 Nano-to-Macro Transport Processes Fall 2004: E HN McaptainhassNo ratings yet
- Introduction To Plasma Physics and Controlled Fusion Notes For Journal 1Document18 pagesIntroduction To Plasma Physics and Controlled Fusion Notes For Journal 1api-272993678100% (1)
- EE 698D Semiconductor Physics Band StructureDocument13 pagesEE 698D Semiconductor Physics Band StructuretransfinitumNo ratings yet
- APhO2001 Theory Prob 2Document2 pagesAPhO2001 Theory Prob 2Popovici DraganNo ratings yet
- Adrian Mulholland Molecular Structure and Bonding (II) SynopsisDocument12 pagesAdrian Mulholland Molecular Structure and Bonding (II) SynopsisAnni bannNo ratings yet
- Lecture 36Document26 pagesLecture 36Imam MashudiNo ratings yet
- Introduction To WavesDocument58 pagesIntroduction To WavesSanu RoyNo ratings yet
- Principles of Molecular SpectrosDocument12 pagesPrinciples of Molecular SpectrosoomganapathiNo ratings yet
- MS200 Chapter 3c Band Theory ShortDocument38 pagesMS200 Chapter 3c Band Theory ShortSwarali GhodkhandeNo ratings yet
- Vibration - Rotation Spectroscopy of HCL and DCLDocument9 pagesVibration - Rotation Spectroscopy of HCL and DCLAngela LamasNo ratings yet
- Vibration-Rotation Spectrum of CO MoleculeDocument10 pagesVibration-Rotation Spectrum of CO MoleculeBishnu RakshitNo ratings yet
- Spectroscopy - Rotational Spectroscopy - Wikiversity PDFDocument36 pagesSpectroscopy - Rotational Spectroscopy - Wikiversity PDFKishore KishoreNo ratings yet
- Spectroscopy - Rotational Spectroscopy - WikiversityDocument36 pagesSpectroscopy - Rotational Spectroscopy - WikiversityKishore KishoreNo ratings yet
- Ch6 - Diatomic Vibration SpectraDocument15 pagesCh6 - Diatomic Vibration SpectraKamsNo ratings yet
- Chapter 4 - Single Particle Motions in Magnetic FieldsDocument32 pagesChapter 4 - Single Particle Motions in Magnetic FieldsDebopam DattaNo ratings yet
- Postgraduate Physics Course on Molecular SpectroscopyDocument18 pagesPostgraduate Physics Course on Molecular SpectroscopyPragya RawalNo ratings yet
- ExcitonsDocument44 pagesExcitonsFei PuNo ratings yet
- Quantum Hall Effect - BDocument7 pagesQuantum Hall Effect - Bspow123No ratings yet
- 01-The Integer Quantum Hall Effect I PDFDocument7 pages01-The Integer Quantum Hall Effect I PDFBheim LlonaNo ratings yet
- Atomic Physics3Document13 pagesAtomic Physics3Rashid AliNo ratings yet
- Single Particle Motion in E and B Fields: 1 ReviewDocument6 pagesSingle Particle Motion in E and B Fields: 1 ReviewLucas Mosimanegape GaileleNo ratings yet
- Ism Chapter 37Document36 pagesIsm Chapter 37NamybiaNo ratings yet
- Vibrational and Rotational Spectroscopy of Diatomic MoleculesDocument6 pagesVibrational and Rotational Spectroscopy of Diatomic MoleculesBharathiNo ratings yet
- Free electron model in metalsDocument8 pagesFree electron model in metalsAntonio RamirezNo ratings yet
- Ch. 5 Inductors, Electromagnets and Permanent MagnetsDocument35 pagesCh. 5 Inductors, Electromagnets and Permanent MagnetslilanxiaNo ratings yet
- ZeemanDocument26 pagesZeemanscrusnNo ratings yet
- APhO2015 Theo Sol1Document9 pagesAPhO2015 Theo Sol1AbhiNo ratings yet
- Structure of Atom Crash CourseDocument98 pagesStructure of Atom Crash CourseghajnisinghoNo ratings yet
- Photochemistry: Prof. M.N.R. Ashfold (S305)Document66 pagesPhotochemistry: Prof. M.N.R. Ashfold (S305)betjodaNo ratings yet
- Rotational Motion: Rotation in Two Dimensions: A Particle On A RingDocument41 pagesRotational Motion: Rotation in Two Dimensions: A Particle On A RingAdelia Ayu WandiraNo ratings yet
- Waves in Media: Ashcroft and Mermin, Solid State Physics (Saunders College, 1976, Page 553)Document42 pagesWaves in Media: Ashcroft and Mermin, Solid State Physics (Saunders College, 1976, Page 553)Amina lbrahimNo ratings yet
- 6 Band Theory of SolidsDocument45 pages6 Band Theory of Solidssaichandrasekhar_dNo ratings yet
- Electronic SpectrosDocument18 pagesElectronic SpectrosjamesNo ratings yet
- Normal Shutdown of BoilerDocument2 pagesNormal Shutdown of BoilerLuqman Sahlan RomadhonaNo ratings yet
- Safety Testing Oh Machines - IFADocument55 pagesSafety Testing Oh Machines - IFAMarco LoiaNo ratings yet
- UV - Vis Spectros PDFDocument25 pagesUV - Vis Spectros PDFNelson BarriosNo ratings yet
- GPMC Profile SummaryDocument51 pagesGPMC Profile SummaryJayadevDamodaranNo ratings yet
- Jagdish Dukre's Guide to Phacoemulsification Machine Components and FunctionsDocument38 pagesJagdish Dukre's Guide to Phacoemulsification Machine Components and FunctionsRizkyAgustriaNo ratings yet
- Light Intensity Control Using Diac and Triac 1Document12 pagesLight Intensity Control Using Diac and Triac 1Prathamesh KaleNo ratings yet
- Teach Yourself Electricity and Electronics 3rd Edition ReviewDocument4 pagesTeach Yourself Electricity and Electronics 3rd Edition ReviewPatricia KalzadaNo ratings yet
- Kvs MagazineDocument88 pagesKvs MagazineGanesh LakshminarasanNo ratings yet
- Answers Exam 21jan 2015Document14 pagesAnswers Exam 21jan 2015Mark HinsterNo ratings yet
- Transformer protection guideDocument195 pagesTransformer protection guideRafael Curiel MedinaNo ratings yet
- Mobile PV Test LabDocument8 pagesMobile PV Test LabMudasir GulzarNo ratings yet
- Smart Grid Communication and Co-Simulation: Vincenzo Liberatore, Member, IEEE Computer, Ahmad Al-HammouriDocument5 pagesSmart Grid Communication and Co-Simulation: Vincenzo Liberatore, Member, IEEE Computer, Ahmad Al-HammouriLahcen AmiriNo ratings yet
- Lista de Articulos: 1 Electrodos Conarco GrupoDocument8 pagesLista de Articulos: 1 Electrodos Conarco GrupoJoel Leandro Ibarra CoriaNo ratings yet
- Nissan Patrol 2007Document22 pagesNissan Patrol 2007GermánNo ratings yet
- Danby Dac6011e Specification SheetDocument1 pageDanby Dac6011e Specification SheetElla MariaNo ratings yet
- Sustainable Power For FutureDocument28 pagesSustainable Power For Futureasim.infniteNo ratings yet
- LOTODocument35 pagesLOTOTopan Mei Cholidin100% (1)
- LabVolt Industrial AC DrivesDocument121 pagesLabVolt Industrial AC Drivesumer farooqNo ratings yet
- Enen 619-03 Final Research ProjectDocument26 pagesEnen 619-03 Final Research ProjectSalman NoorNo ratings yet
- Athanassios A. Nassikas - The Notion of Aether As A Possible Consequence of A Claim For Minimum ContradictionsDocument12 pagesAthanassios A. Nassikas - The Notion of Aether As A Possible Consequence of A Claim For Minimum ContradictionsKunma050No ratings yet
- Problems Worksheet: Specific Heat CapacityDocument5 pagesProblems Worksheet: Specific Heat CapacityDilini WijesinghNo ratings yet
- Liquid ElectrictyDocument17 pagesLiquid Electrictysrikanthdude100% (1)
- TA7270 PDocument11 pagesTA7270 PsuzukkNo ratings yet
- CNWX PDFDocument6 pagesCNWX PDFNghi VoNo ratings yet
- 815 FujianDocument68 pages815 FujianHammami SalahNo ratings yet
- Team Tec IncineratorDocument184 pagesTeam Tec IncineratorRobinson SoledadNo ratings yet
- Carbondioxide in Water EquilibriumDocument7 pagesCarbondioxide in Water EquilibriumsaaroomaniNo ratings yet
- ULSD-Lubrcity Spike-Nom-16-EN590Document3 pagesULSD-Lubrcity Spike-Nom-16-EN590Angel LealNo ratings yet