You might also like
- Cystic Fibrosis Diagnosis, Treatment & ManagementDocument28 pagesCystic Fibrosis Diagnosis, Treatment & ManagementAnita ThompsonNo ratings yet
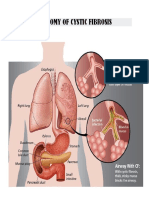
- Anatomy of Cystic FibrosisDocument5 pagesAnatomy of Cystic FibrosislazylawatudentNo ratings yet
- Cystic Fibrosis Final AssignmentDocument6 pagesCystic Fibrosis Final AssignmentSteg250No ratings yet
- Quiz 1Document3 pagesQuiz 1Wiljohn de la CruzNo ratings yet
- Ymphatic Ystem: Natomy AND HysiologyDocument7 pagesYmphatic Ystem: Natomy AND Hysiologydlneisha61No ratings yet
- Running Head: Reverse Case Study 1Document15 pagesRunning Head: Reverse Case Study 1muthegetra100% (1)
- Case Press Medical WardDocument29 pagesCase Press Medical WardivanloveschieNo ratings yet
- Disseminated Intravascular CoagulationDocument2 pagesDisseminated Intravascular CoagulationVince100% (1)
- Anatomy and Phsyiology of MeningococcemiaDocument2 pagesAnatomy and Phsyiology of MeningococcemiaKevin Comahig100% (1)
- Nursing Ethics DilemmasDocument3 pagesNursing Ethics DilemmasDeng Navarro PasionNo ratings yet
- Understanding Amyotrophic Lateral Sclerosis (ALSDocument17 pagesUnderstanding Amyotrophic Lateral Sclerosis (ALSJoel Andrew Java Daga100% (2)
- Assignment 4Document8 pagesAssignment 4api-328441669100% (1)
- Oxygen Therapy For Chronic Obstructive Pulmonary Disease PatientsDocument9 pagesOxygen Therapy For Chronic Obstructive Pulmonary Disease PatientsshielphiNo ratings yet
- Final Thesis With CV (For Bookbind)Document138 pagesFinal Thesis With CV (For Bookbind)Bea Marie Eclevia100% (1)
- COPD: Chronic Obstructive Pulmonary DiseaseDocument3 pagesCOPD: Chronic Obstructive Pulmonary DiseaseRichard TanNo ratings yet
- Acute Respiratory Distress Syndrome (ARDS): Causes, Symptoms and TreatmentDocument30 pagesAcute Respiratory Distress Syndrome (ARDS): Causes, Symptoms and TreatmentOya Zuraini KamalNo ratings yet
- Gordons TypologyDocument37 pagesGordons TypologyJo PigarNo ratings yet
- Can We Be Good Without God? Morality Requires an Absolute Moral Law GiverDocument1 pageCan We Be Good Without God? Morality Requires an Absolute Moral Law GiverRaphael SisonNo ratings yet
- NCM116 Module 3 Cerebrovascular disease QuizDocument5 pagesNCM116 Module 3 Cerebrovascular disease QuizIvan FernandezNo ratings yet
- Rabies-Complete Report - San Lazaro HospitalDocument13 pagesRabies-Complete Report - San Lazaro HospitalMichelle Palacio-De la Cruz100% (1)
- Pott Disease 1223292121651385 8Document54 pagesPott Disease 1223292121651385 8Ismail SalimNo ratings yet
- Assessment of Respiratory SystemDocument34 pagesAssessment of Respiratory SystemKristal Jade Yanto Esquillo100% (1)
- Man, Health, & Illness: InsertDocument50 pagesMan, Health, & Illness: InsertJan Oliver YaresNo ratings yet
- 04 - Typhoid FeverDocument35 pages04 - Typhoid Feversoheil100% (1)
- BioethicsDocument3 pagesBioethicspaui16No ratings yet
- Congenital HypothyroidismDocument37 pagesCongenital HypothyroidismAwaluddin SalamNo ratings yet
- Pneumococcal PneumoniaDocument13 pagesPneumococcal PneumoniaRae Marie AquinoNo ratings yet
- Acute Glomerulonephritis (AGN) : Group A Beta Hemolytic StretococcusDocument3 pagesAcute Glomerulonephritis (AGN) : Group A Beta Hemolytic StretococcusKristine Danielle DejeloNo ratings yet
- Health and Wellness ConceptsDocument38 pagesHealth and Wellness ConceptsKathrina AlfonsoNo ratings yet
- Cystic Fibrosis - Patient PresentationDocument24 pagesCystic Fibrosis - Patient Presentationapi-396515657No ratings yet
- Riehl's Self-Concept Module of NursingDocument1 pageRiehl's Self-Concept Module of NursingRachelle Geronimo100% (1)
- Dengue Fever: Causes, Symptoms and PreventionDocument7 pagesDengue Fever: Causes, Symptoms and PreventionAmber Hope PonsicaNo ratings yet
- DYSTROPHYDocument15 pagesDYSTROPHYleeyan2wenty6No ratings yet
- NCM 103 Fundamentals of NursingDocument43 pagesNCM 103 Fundamentals of NursingYzobel Phoebe ParoanNo ratings yet
- Cystic Fibrosis PathophysiologyDocument5 pagesCystic Fibrosis PathophysiologyKim Enrico JumarangNo ratings yet
- Hepatic EncephalopathyDocument3 pagesHepatic EncephalopathyAnonymous GIGXKjfLNo ratings yet
- Expanded Program On ImmunizationDocument4 pagesExpanded Program On ImmunizationcharmdoszNo ratings yet
- Polycythemia Guide: Causes, Symptoms & TreatmentDocument16 pagesPolycythemia Guide: Causes, Symptoms & TreatmentVanessa Camille DomingoNo ratings yet
- 7 - Regulation and Functions of The Thyroid HormonesDocument31 pages7 - Regulation and Functions of The Thyroid HormonesVigneshwaran RavishankarNo ratings yet
- 780 Adult Cardio Resp Assess DSTDocument10 pages780 Adult Cardio Resp Assess DSTGursangeet Kaur100% (1)
- Fetal CirculationDocument2 pagesFetal CirculationLhovely ConwiNo ratings yet
- Decent Work Country Profile PHILIPPINESDocument118 pagesDecent Work Country Profile PHILIPPINESi_am_alvinNo ratings yet
- Commission On Higher Education Center of Excellence in Nursing EducationDocument9 pagesCommission On Higher Education Center of Excellence in Nursing EducationR-Chian Jose GermanpNo ratings yet
- The Infectious ProcessDocument4 pagesThe Infectious ProcessAdnan Akram, MD (Latvia)100% (1)
- Human Immunodeficiency Virus Case AnalysisDocument5 pagesHuman Immunodeficiency Virus Case AnalysisAllen Bugarin CabadingNo ratings yet
- Common Health Problems of Infancy PowptDocument78 pagesCommon Health Problems of Infancy PowptCiella Dela CruzNo ratings yet
- Glasgo Coma ScaleDocument3 pagesGlasgo Coma ScaleMichael LinebargerNo ratings yet
- Kidney Transplantation: Group 9 Syazwaniyati Nurulasmira Nur Hamiza Siti NursuhadaDocument12 pagesKidney Transplantation: Group 9 Syazwaniyati Nurulasmira Nur Hamiza Siti NursuhadaSiti Nursuhada binti Mohd AminNo ratings yet
- ICU Case Study PDFDocument19 pagesICU Case Study PDFYashvi SinghNo ratings yet
- Burns MGNT and CalculationDocument7 pagesBurns MGNT and CalculationPoova RagavanNo ratings yet
- HA-RLE-WS # 5 Assessing General Status and Vital SignsDocument5 pagesHA-RLE-WS # 5 Assessing General Status and Vital SignsJULIE ANNE CORTEZ100% (1)
- Philippine Nursing Law of 2002 Ra # 9173Document15 pagesPhilippine Nursing Law of 2002 Ra # 9173Michael Maunda AmpuanNo ratings yet
- Seminar On: Cystic FibrosisDocument39 pagesSeminar On: Cystic Fibrosislumina.s100% (1)
- On the Inviolability of Human Life: Julia Kristeva's Reflections on the Universal Abolition of the Death PenaltyDocument6 pagesOn the Inviolability of Human Life: Julia Kristeva's Reflections on the Universal Abolition of the Death PenaltyEunbit YiNo ratings yet
- Review of SystemsDocument4 pagesReview of SystemsKat ArriolaNo ratings yet
- Care of Older Person with Chronic IllnessDocument87 pagesCare of Older Person with Chronic IllnessKristine KimNo ratings yet
- Immunodeficiency DisordersDocument8 pagesImmunodeficiency Disordersbpt2No ratings yet
- Cellular AberrationsDocument94 pagesCellular AberrationsridzkhaNo ratings yet
- Chapter 7 Cystic FibrosisDocument9 pagesChapter 7 Cystic FibrosisMariana DariiNo ratings yet
- EportfolioDocument7 pagesEportfolioapi-344036469No ratings yet
- Cystic Fibrosis: SymptomsDocument2 pagesCystic Fibrosis: SymptomsMehrul Singh RanavatNo ratings yet
- NCP Cystic Fibrosis (Example)Document3 pagesNCP Cystic Fibrosis (Example)Lizet QuiamcoNo ratings yet
- Cystic Fibrosis 1Document7 pagesCystic Fibrosis 1api-306098893No ratings yet
- Pedia Disorders Upgraded For Lnu CaDocument6 pagesPedia Disorders Upgraded For Lnu Cashirlenedel cariñoNo ratings yet
- Respiratory MCQDocument10 pagesRespiratory MCQSyeda Aroosa Abbas Naqvi100% (1)
- Pnle July 2013 DX Test (Np3) III ANSWER PRINTDocument29 pagesPnle July 2013 DX Test (Np3) III ANSWER PRINTPaul EspinosaNo ratings yet
- Chronic Diarrhea in ChildrenDocument4 pagesChronic Diarrhea in ChildrenDaffa Ahmad NaufalNo ratings yet
- AKP Jan. 2022Document30 pagesAKP Jan. 2022jcNo ratings yet
- Ch. 11 Complex Inheritance and Human Heredity NotesDocument28 pagesCh. 11 Complex Inheritance and Human Heredity NotesMaya KhaledNo ratings yet
- GEN2MHG Notes Part 3Document26 pagesGEN2MHG Notes Part 3studycosmilkNo ratings yet
- Cystic FibrosisDocument32 pagesCystic FibrosisSrm Genetics100% (1)
- Hypernatremia: S. Faubel and J. Topf 8 HypernatremiaDocument42 pagesHypernatremia: S. Faubel and J. Topf 8 HypernatremiaJoel Topf100% (2)
- AHCCCS Pharmacy and Therapeutics Committee May 19, 2021Document247 pagesAHCCCS Pharmacy and Therapeutics Committee May 19, 2021hussam mazinNo ratings yet
- Disorder Cause Treatment/Possible TreatmentDocument3 pagesDisorder Cause Treatment/Possible TreatmentNoval, SheanNo ratings yet
- Case Study 103Document8 pagesCase Study 103Jonah MaasinNo ratings yet
- Maths For Biology Questions and Answers AQA OCR EdexcelDocument15 pagesMaths For Biology Questions and Answers AQA OCR EdexcelsciencedocsmanNo ratings yet
- MBT-Term Test 1Document45 pagesMBT-Term Test 1lkokodkodNo ratings yet
- Chapter 10 - Diseases of Infancy and ChildhoodDocument17 pagesChapter 10 - Diseases of Infancy and ChildhoodAgnieszka WisniewskaNo ratings yet
- USMLE Questions SummarizedDocument85 pagesUSMLE Questions SummarizedJamesIwu89% (19)
- PBL Project Planning Guide Cell MembraneDocument2 pagesPBL Project Planning Guide Cell MembraneJohn OsborneNo ratings yet
- Rekayasa Genetik, Konsultasi Genetik Dan Terapi GeneDocument124 pagesRekayasa Genetik, Konsultasi Genetik Dan Terapi GeneRollyRiksantoNo ratings yet
- (Advances in Experimental Medicine and Biology 931) Christine Imbert (Eds.)-Fungal Biofilms and Related Infections_ Advances in Microbiology, Infectious Diseases and Public Health Volume 3-Springer InDocument129 pages(Advances in Experimental Medicine and Biology 931) Christine Imbert (Eds.)-Fungal Biofilms and Related Infections_ Advances in Microbiology, Infectious Diseases and Public Health Volume 3-Springer InVerónica Duménez JofréNo ratings yet
- Practice Assignment 4 - Solutions - 2012Document13 pagesPractice Assignment 4 - Solutions - 2012jeankerlensNo ratings yet
- D - 5answer KeyDocument15 pagesD - 5answer KeyJune DumdumayaNo ratings yet
- Micropara LabDocument8 pagesMicropara LabFatima KateNo ratings yet
- NCM 116 Medical SurgicalDocument9 pagesNCM 116 Medical SurgicalIvan A. EleginoNo ratings yet
- Booklet The Structures of LifeDocument66 pagesBooklet The Structures of LifeIrving EuanNo ratings yet
- Pseudomonas Biofilm Formation and Antibiotic Resistance Are Linked To Phenotypic VariationDocument4 pagesPseudomonas Biofilm Formation and Antibiotic Resistance Are Linked To Phenotypic VariationChadi AzarNo ratings yet
- CF Guideline 2011 - Jan 4Document200 pagesCF Guideline 2011 - Jan 4vtNo ratings yet
- Diabetes Classification UpdateDocument241 pagesDiabetes Classification UpdateAlex MarinNo ratings yet
- Why We Die: The New Science of Aging and the Quest for ImmortalityFrom EverandWhy We Die: The New Science of Aging and the Quest for ImmortalityRating: 4 out of 5 stars4/5 (3)
- The Age of Magical Overthinking: Notes on Modern IrrationalityFrom EverandThe Age of Magical Overthinking: Notes on Modern IrrationalityRating: 4 out of 5 stars4/5 (15)
- Think This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeFrom EverandThink This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeNo ratings yet
- Summary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedFrom EverandSummary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedRating: 5 out of 5 stars5/5 (78)
- The Obesity Code: Unlocking the Secrets of Weight LossFrom EverandThe Obesity Code: Unlocking the Secrets of Weight LossRating: 5 out of 5 stars5/5 (4)
- LIT: Life Ignition Tools: Use Nature's Playbook to Energize Your Brain, Spark Ideas, and Ignite ActionFrom EverandLIT: Life Ignition Tools: Use Nature's Playbook to Energize Your Brain, Spark Ideas, and Ignite ActionRating: 4 out of 5 stars4/5 (402)
- Raising Mentally Strong Kids: How to Combine the Power of Neuroscience with Love and Logic to Grow Confident, Kind, Responsible, and Resilient Children and Young AdultsFrom EverandRaising Mentally Strong Kids: How to Combine the Power of Neuroscience with Love and Logic to Grow Confident, Kind, Responsible, and Resilient Children and Young AdultsRating: 5 out of 5 stars5/5 (1)
- The Ritual Effect: From Habit to Ritual, Harness the Surprising Power of Everyday ActionsFrom EverandThe Ritual Effect: From Habit to Ritual, Harness the Surprising Power of Everyday ActionsRating: 3.5 out of 5 stars3.5/5 (3)
- By the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsFrom EverandBy the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsNo ratings yet
- The Ultimate Guide To Memory Improvement TechniquesFrom EverandThe Ultimate Guide To Memory Improvement TechniquesRating: 5 out of 5 stars5/5 (34)
- Roxane Gay & Everand Originals: My Year of Psychedelics: Lessons on Better LivingFrom EverandRoxane Gay & Everand Originals: My Year of Psychedelics: Lessons on Better LivingRating: 3.5 out of 5 stars3.5/5 (33)
- Raising Good Humans: A Mindful Guide to Breaking the Cycle of Reactive Parenting and Raising Kind, Confident KidsFrom EverandRaising Good Humans: A Mindful Guide to Breaking the Cycle of Reactive Parenting and Raising Kind, Confident KidsRating: 4.5 out of 5 stars4.5/5 (169)
- Techniques Exercises And Tricks For Memory ImprovementFrom EverandTechniques Exercises And Tricks For Memory ImprovementRating: 4.5 out of 5 stars4.5/5 (40)
- Dark Psychology & Manipulation: Discover How To Analyze People and Master Human Behaviour Using Emotional Influence Techniques, Body Language Secrets, Covert NLP, Speed Reading, and Hypnosis.From EverandDark Psychology & Manipulation: Discover How To Analyze People and Master Human Behaviour Using Emotional Influence Techniques, Body Language Secrets, Covert NLP, Speed Reading, and Hypnosis.Rating: 4.5 out of 5 stars4.5/5 (110)
- Summary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisFrom EverandSummary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisRating: 4.5 out of 5 stars4.5/5 (42)
- The Courage Habit: How to Accept Your Fears, Release the Past, and Live Your Courageous LifeFrom EverandThe Courage Habit: How to Accept Your Fears, Release the Past, and Live Your Courageous LifeRating: 4.5 out of 5 stars4.5/5 (253)
- The Body Keeps the Score by Bessel Van der Kolk, M.D. - Book Summary: Brain, Mind, and Body in the Healing of TraumaFrom EverandThe Body Keeps the Score by Bessel Van der Kolk, M.D. - Book Summary: Brain, Mind, and Body in the Healing of TraumaRating: 4.5 out of 5 stars4.5/5 (266)
- Roxane Gay & Everand Originals: My Year of Psychedelics: Lessons on Better LivingFrom EverandRoxane Gay & Everand Originals: My Year of Psychedelics: Lessons on Better LivingRating: 5 out of 5 stars5/5 (5)
- Mindset by Carol S. Dweck - Book Summary: The New Psychology of SuccessFrom EverandMindset by Carol S. Dweck - Book Summary: The New Psychology of SuccessRating: 4.5 out of 5 stars4.5/5 (328)
- Outlive: The Science and Art of Longevity by Peter Attia: Key Takeaways, Summary & AnalysisFrom EverandOutlive: The Science and Art of Longevity by Peter Attia: Key Takeaways, Summary & AnalysisRating: 4 out of 5 stars4/5 (1)
- The Happiness Trap: How to Stop Struggling and Start LivingFrom EverandThe Happiness Trap: How to Stop Struggling and Start LivingRating: 4 out of 5 stars4/5 (1)
- Cult, A Love Story: Ten Years Inside a Canadian Cult and the Subsequent Long Road of RecoveryFrom EverandCult, A Love Story: Ten Years Inside a Canadian Cult and the Subsequent Long Road of RecoveryRating: 4 out of 5 stars4/5 (44)
- The Tennis Partner: A Doctor's Story of Friendship and LossFrom EverandThe Tennis Partner: A Doctor's Story of Friendship and LossRating: 4.5 out of 5 stars4.5/5 (4)