You might also like
- Biochemistry - A Short CourseDocument1,026 pagesBiochemistry - A Short CourseFelicita Urzi100% (3)
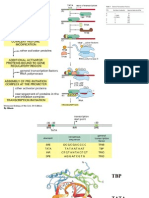
- TRP OperonDocument80 pagesTRP OperonIsmi SitiNo ratings yet
- CH 10 Gene ExpressionDocument23 pagesCH 10 Gene ExpressionerichaasNo ratings yet
- Gene Regulation: Mrs. Ofelia Solano SaludarDocument39 pagesGene Regulation: Mrs. Ofelia Solano SaludarmskikiNo ratings yet
- Fat-Soluble Vitamins A, D, E & K: Roles, Sources & ImportanceDocument33 pagesFat-Soluble Vitamins A, D, E & K: Roles, Sources & ImportanceDianzerShengNo ratings yet
- Gene Regulation in Prokaryotes-1Document17 pagesGene Regulation in Prokaryotes-1Fasiha Mushadi100% (1)
- SOLVE THE CRIME OF THE FIRE-BREATHING MUTANTDocument2 pagesSOLVE THE CRIME OF THE FIRE-BREATHING MUTANTeula faith miracle andamNo ratings yet
- PCR Questions and Answers GuideDocument5 pagesPCR Questions and Answers GuideMohita ChughNo ratings yet
- Tarif Prodia 2020 Untuk EmailDocument32 pagesTarif Prodia 2020 Untuk Emailklinik bhaksenatragia100% (2)
- RI TYS Ans 2007-2019-CompressedDocument215 pagesRI TYS Ans 2007-2019-CompressedelgiNo ratings yet
- Watson RegulaciónDocument45 pagesWatson RegulaciónsergioNo ratings yet
- Fis Upstream BindingDocument15 pagesFis Upstream BindingDiegoNo ratings yet
- Lecture Transcriptional Control in Prokaryotes and EukaryotesDocument13 pagesLecture Transcriptional Control in Prokaryotes and EukaryotesTony_Tonique_Q_1649No ratings yet
- BMC BiotechnologyDocument12 pagesBMC BiotechnologymamlikatuNo ratings yet
- 3-Nucleic Acids Research - 2020 - Crespo I Et, AlDocument15 pages3-Nucleic Acids Research - 2020 - Crespo I Et, AlAntonNo ratings yet
- Epigenetics Role in Alternative RNA SplicingDocument11 pagesEpigenetics Role in Alternative RNA SplicingThiagoNo ratings yet
- 01 - Doniselli Et Al. 2015Document14 pages01 - Doniselli Et Al. 2015Edgar Huerta CardenasNo ratings yet
- Chap 15Document16 pagesChap 15araneyaNo ratings yet
- Transcription FactorsDocument25 pagesTranscription FactorsPriya.RNo ratings yet
- Bio CH 18 OutlineDocument3 pagesBio CH 18 OutlineDee MarNo ratings yet
- Pembuatan Nata de CocoDocument4 pagesPembuatan Nata de CocoRahayufirniaNo ratings yet
- Regulación Por AtenuaciónDocument5 pagesRegulación Por AtenuaciónKaryme Aylín Martinez OliverosNo ratings yet
- TECHNIQUES IN MOLECULAR BIOLOGY – PROMOTERS AND PLASMIDSDocument4 pagesTECHNIQUES IN MOLECULAR BIOLOGY – PROMOTERS AND PLASMIDSAndika MardiantoNo ratings yet
- Piggybac Transposase and Transposon Derivatives For Gene Transfer Targeting The Ribosomal Dna Loci of Cho CellsDocument9 pagesPiggybac Transposase and Transposon Derivatives For Gene Transfer Targeting The Ribosomal Dna Loci of Cho CellsHonesty AnandaNo ratings yet
- Quantitative Northern Blot Analysis of Mammalian rRNA ProcessingDocument11 pagesQuantitative Northern Blot Analysis of Mammalian rRNA ProcessingAlejandro NarvaezNo ratings yet
- Miguel-Arribas A, Et Al. NAR 2021Document15 pagesMiguel-Arribas A, Et Al. NAR 2021AntonNo ratings yet
- PMC4027209 Gku180Document10 pagesPMC4027209 Gku180Gotham BuddhaNo ratings yet
- Decker 1Document9 pagesDecker 1Katarina Kaca GacevicNo ratings yet
- Methods For Alternative SplicingDocument16 pagesMethods For Alternative SplicingRaji SivarupaNo ratings yet
- Expression Vectors 1Document15 pagesExpression Vectors 1keyonthewebNo ratings yet
- Transcription FactorsDocument32 pagesTranscription FactorsParijat BanerjeeNo ratings yet
- Ghaz-Jahanian2013 Article InfluenceOfSmallRNAsOnBiofilmFDocument10 pagesGhaz-Jahanian2013 Article InfluenceOfSmallRNAsOnBiofilmFpapahojaloveNo ratings yet
- Transcription Is A Cellular Process During Which RNA Is SynthesizedDocument7 pagesTranscription Is A Cellular Process During Which RNA Is SynthesizedDeborah ChemutaiNo ratings yet
- Ekspresi Gen PDFDocument12 pagesEkspresi Gen PDFRose DesyNo ratings yet
- Control of SiRNA Expression Using The CreloxP Recombination SystemDocument8 pagesControl of SiRNA Expression Using The CreloxP Recombination SystemGaurav GyanwaliNo ratings yet
- Unit 3Document33 pagesUnit 3Dsce BtNo ratings yet
- RNA SplicingDocument14 pagesRNA SplicingnurlianaNo ratings yet
- AP Q Chapter 4Document56 pagesAP Q Chapter 4Mahra AlketbiNo ratings yet
- Tarnscription RNA POLYMERASEDocument26 pagesTarnscription RNA POLYMERASEnarasimmareddy.cNo ratings yet
- CTCF Regulates The Local Epigenetic State of Ribosomal DNA RepeatsDocument21 pagesCTCF Regulates The Local Epigenetic State of Ribosomal DNA RepeatsPaige MunroeNo ratings yet
- Transcription Prokaryotes 2012-cDocument27 pagesTranscription Prokaryotes 2012-cAnupama PatiNo ratings yet
- Alternative Splicing in PlantsDocument8 pagesAlternative Splicing in Plantsanon_442538974No ratings yet
- The Influence of Silent Mutations On Pre-mRNA SplicingDocument24 pagesThe Influence of Silent Mutations On Pre-mRNA SplicingAtikaWidyaSyariNo ratings yet
- Genetics of ProkaryotesDocument2 pagesGenetics of ProkaryotesReyes ChengNo ratings yet
- Transposition of Native Chromatin For Fast and Sensitive Epigenomic Profiling of Open Chromatin, Dna-Binding Proteins and Nucleosome PositionDocument8 pagesTransposition of Native Chromatin For Fast and Sensitive Epigenomic Profiling of Open Chromatin, Dna-Binding Proteins and Nucleosome PositionKito TongHuiNo ratings yet
- BIO230 - Section 1 Regulation of Genome Expression Lecture 1-9Document36 pagesBIO230 - Section 1 Regulation of Genome Expression Lecture 1-9yusrawasim147No ratings yet
- Proud Foot 2002Document12 pagesProud Foot 2002Mumun Hal MuratNo ratings yet
- Gene Regulation (Post Translation)Document111 pagesGene Regulation (Post Translation)raryanraj44No ratings yet
- Gene Regulation in Prokaryotes: Presented by Dr. Nitin M. Kamble PHD Scholar BtyDocument33 pagesGene Regulation in Prokaryotes: Presented by Dr. Nitin M. Kamble PHD Scholar BtyNitin KambleNo ratings yet
- 2-Nucleic Acids Research - 2020 - Singh PK Et, AlDocument17 pages2-Nucleic Acids Research - 2020 - Singh PK Et, AlAntonNo ratings yet
- D Mallika ComputerscienceDocument62 pagesD Mallika ComputerscienceDhilsanth SLNo ratings yet
- Ap BioDocument13 pagesAp BiomenoahisloveNo ratings yet
- Termination and Regulation of Bacterial TranscriptionDocument95 pagesTermination and Regulation of Bacterial TranscriptionhaiduvnNo ratings yet
- Order ID 406406482 Gene ExpressionDocument4 pagesOrder ID 406406482 Gene ExpressionjohnessamburuNo ratings yet
- Functional GenomicsDocument11 pagesFunctional Genomicswatson191No ratings yet
- Meredith Barb Homework-2 (25 Points) Due On Sept. 24 by 5:00pmDocument5 pagesMeredith Barb Homework-2 (25 Points) Due On Sept. 24 by 5:00pmMeredith BarbNo ratings yet
- PP3_2009_Dorsch_Nature Methods_Analysis of receptor oligomerization by FRAP microscopyDocument6 pagesPP3_2009_Dorsch_Nature Methods_Analysis of receptor oligomerization by FRAP microscopydamon tanNo ratings yet
- Gene Expression Regulation by Transcription Attenuation MechanismsDocument12 pagesGene Expression Regulation by Transcription Attenuation MechanismsazzaassNo ratings yet
- TMP FD75Document11 pagesTMP FD75FrontiersNo ratings yet
- Assignment On Regulation of Transcription, Lytic Lysogeny Cascade and SOS Regulatory SystemDocument16 pagesAssignment On Regulation of Transcription, Lytic Lysogeny Cascade and SOS Regulatory SystemShraddha Bhatt ChavanNo ratings yet
- (Full) Transcriptional Regulation in Prokaryotes - Alper K.Document15 pages(Full) Transcriptional Regulation in Prokaryotes - Alper K.Simay BoztaşNo ratings yet
- Gateway Technology: M.Phil. Biotechnology CBM, University of SwatDocument9 pagesGateway Technology: M.Phil. Biotechnology CBM, University of SwatGul AfshaNo ratings yet
- Transcription Factor Dynamics Mini ReviewDocument8 pagesTranscription Factor Dynamics Mini ReviewwebhappyNo ratings yet
- Molecular Basis of Inheritance 2Document6 pagesMolecular Basis of Inheritance 2VARSHAN GAMINGNo ratings yet
- Gene Regulation by The Act of Long Non-Coding RNADocument14 pagesGene Regulation by The Act of Long Non-Coding RNAMarina Célia Nunes Ferreira Da C SilvaNo ratings yet
- Lab 1 - Text Processing in Linux N-GramsDocument8 pagesLab 1 - Text Processing in Linux N-GramsAlberto Mario Ceballos ArroyoNo ratings yet
- Solarenergy spa-CODocument26 pagesSolarenergy spa-COAlberto Mario Ceballos ArroyoNo ratings yet
- Ejercicio de Traducción Con Enfoque Comunicativo ChristianDocument4 pagesEjercicio de Traducción Con Enfoque Comunicativo ChristianAlberto Mario Ceballos ArroyoNo ratings yet
- Subiect Admitere 2018 Iulie InformaticaDocument16 pagesSubiect Admitere 2018 Iulie InformaticaOanaPavelNo ratings yet
- Lecture6 Levelsets3 PDFDocument59 pagesLecture6 Levelsets3 PDFAlberto Mario Ceballos ArroyoNo ratings yet
- RamdomForest Tolima OpticaSARDocument3 pagesRamdomForest Tolima OpticaSARAlberto Mario Ceballos ArroyoNo ratings yet
- Nominados A Las Becas Fulbright - Convocatoria 2017, Cohorte 2018 Beca Fulbright - Saldarriaga ConchaDocument3 pagesNominados A Las Becas Fulbright - Convocatoria 2017, Cohorte 2018 Beca Fulbright - Saldarriaga ConchaAlberto Mario Ceballos ArroyoNo ratings yet
- PreguntasDocument1 pagePreguntasAlberto Mario Ceballos ArroyoNo ratings yet
- ArenasP - HerreraS - NoreñaV - QuirozL SlidesDocument28 pagesArenasP - HerreraS - NoreñaV - QuirozL SlidesAlberto Mario Ceballos ArroyoNo ratings yet
- PD Dr.-Ing. Lena Maier-Hein's Curriculum VitaeDocument5 pagesPD Dr.-Ing. Lena Maier-Hein's Curriculum VitaeAlberto Mario Ceballos ArroyoNo ratings yet
- Designing LED Array For Uniform Illumination Distribution by Simulated Annealing AlgorithmDocument13 pagesDesigning LED Array For Uniform Illumination Distribution by Simulated Annealing AlgorithmAlberto Mario Ceballos ArroyoNo ratings yet
- Evolution and The Illusion of Randomness: Stephen L. TalbottDocument28 pagesEvolution and The Illusion of Randomness: Stephen L. TalbottAlberto Mario Ceballos ArroyoNo ratings yet
- Matrixcookbook PDFDocument72 pagesMatrixcookbook PDFeetahaNo ratings yet
- PD Dr.-Ing. Lena Maier-Hein's Curriculum VitaeDocument5 pagesPD Dr.-Ing. Lena Maier-Hein's Curriculum VitaeAlberto Mario Ceballos ArroyoNo ratings yet
- An Overview of Color Constancy Algorithms: June 2006Document14 pagesAn Overview of Color Constancy Algorithms: June 2006Alberto Mario Ceballos ArroyoNo ratings yet
- Color Correction Matrix For Sparse RGB-W Image Sensor Without IR Cutoff Filter PDFDocument11 pagesColor Correction Matrix For Sparse RGB-W Image Sensor Without IR Cutoff Filter PDFAlberto Mario Ceballos ArroyoNo ratings yet
- Matrix CalculusDocument9 pagesMatrix CalculusWmleaoNo ratings yet
- Sanger SequencingDocument4 pagesSanger SequencingMilka RahmanNo ratings yet
- DNA Fingerprinting ActivityDocument2 pagesDNA Fingerprinting ActivityCaryl Louise ParlanNo ratings yet
- ZOO 114 DNA Function LectureDocument53 pagesZOO 114 DNA Function Lecturerakingbulugbe194No ratings yet
- Edited Activity 1 8 2Document2 pagesEdited Activity 1 8 2Karl Mathew PajarillagaNo ratings yet
- Amino Acids and ProteinDocument32 pagesAmino Acids and ProteinArchishmaan UdgataNo ratings yet
- Biochemistry Final Exam Preview Guide - 2019Document3 pagesBiochemistry Final Exam Preview Guide - 2019jake0% (1)
- PhotosynthesisDocument23 pagesPhotosynthesisSandy IkbarNo ratings yet
- Biological Molecules 2Document9 pagesBiological Molecules 2Feranmi AkinboboyeNo ratings yet
- Lipids Midterm NotesDocument10 pagesLipids Midterm NotesJBNo ratings yet
- CH407 Biochemical EngineeringDocument2 pagesCH407 Biochemical EngineeringVish VishwasNo ratings yet
- Nume, Prenume Domiciliu Varsta Sex M/FDocument38 pagesNume, Prenume Domiciliu Varsta Sex M/FAndreea BarizNo ratings yet
- DPN - Biochem 1-Exam 2 - 2022Document9 pagesDPN - Biochem 1-Exam 2 - 2022chienyu2002No ratings yet
- PROM-21949-001 - TD - QCC Protocol Web-Overview - Ver3 - 0323 - WWDocument12 pagesPROM-21949-001 - TD - QCC Protocol Web-Overview - Ver3 - 0323 - WWNabil FIKRAOUINo ratings yet
- Human Molecular Genetics: Fourth EditionDocument67 pagesHuman Molecular Genetics: Fourth EditionMousumi BalNo ratings yet
- Granular Endoplasmic ReticulumDocument2 pagesGranular Endoplasmic ReticulumSetumil PowerNo ratings yet
- MRCP 1 Masterpass AnswersDocument43 pagesMRCP 1 Masterpass AnswersMJwesleyNo ratings yet
- Chapter 25 - Synthetic and Natural Organic PolymersDocument14 pagesChapter 25 - Synthetic and Natural Organic Polymersmaniz442100% (1)
- Gizi Vitamin EDocument97 pagesGizi Vitamin EMaya DasmaselaNo ratings yet
- Operons: How Bacteria Regulate Gene ExpressionDocument3 pagesOperons: How Bacteria Regulate Gene ExpressionS NairNo ratings yet
- Mechanisms of Long Noncoding RNA Function in Development and DiseaseDocument19 pagesMechanisms of Long Noncoding RNA Function in Development and DiseaseManuel HernándezNo ratings yet
- Tutorial 5 - Lipid Metabolism Tutorial CaseDocument8 pagesTutorial 5 - Lipid Metabolism Tutorial CaseSofíaGriggsNo ratings yet
- Biology 3-20 SecAss 12-1Document1 pageBiology 3-20 SecAss 12-1Edward Egan100% (1)
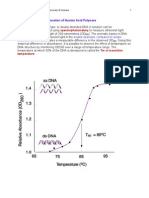
- Factors Affecting DNA Denaturation and RenaturationDocument4 pagesFactors Affecting DNA Denaturation and RenaturationVenkateswarlu Yadavalli100% (2)
- Enzymology SummaryDocument6 pagesEnzymology SummaryRyan Fortune AludaNo ratings yet