You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5811)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1092)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (844)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (897)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (540)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (348)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (822)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (401)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Minimum Design Metal Temperature PresentationDocument21 pagesMinimum Design Metal Temperature PresentationAleck Vieyra100% (1)
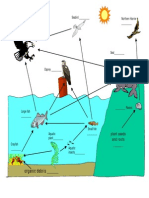
- Foodweb KeyDocument3 pagesFoodweb KeylsueyinNo ratings yet
- C++ ExerciseDocument49 pagesC++ ExerciselsueyinNo ratings yet
- Zeolites and Zeolite Like Material in Industrial CatalysisDocument33 pagesZeolites and Zeolite Like Material in Industrial CatalysislsueyinNo ratings yet
- What Are Porous MaterialsDocument33 pagesWhat Are Porous MaterialslsueyinNo ratings yet
- Organic Photochemistry: Prof. Dr. Burkhard König, Institut Für Organische Chemie, Universität RegensburgDocument38 pagesOrganic Photochemistry: Prof. Dr. Burkhard König, Institut Für Organische Chemie, Universität RegensburglsueyinNo ratings yet
- Ion-Selective Electrode Determination of Fluoride Ion: Chemistry 321L ManualDocument5 pagesIon-Selective Electrode Determination of Fluoride Ion: Chemistry 321L Manuallsueyin100% (1)
- Towards The Rational Synthesis of ZeolitesDocument0 pagesTowards The Rational Synthesis of ZeoliteslsueyinNo ratings yet
- Disperse Systems. The Methods of Preparing of Colloidal Solutions. Their PropertiesDocument33 pagesDisperse Systems. The Methods of Preparing of Colloidal Solutions. Their Propertieslsueyin100% (1)
- MOF NoteDocument61 pagesMOF NotelsueyinNo ratings yet
- ERT 313 Bioseparation Engineering Adsorption: Prepared By: Miss Hairul Nazirah Abdul HalimDocument25 pagesERT 313 Bioseparation Engineering Adsorption: Prepared By: Miss Hairul Nazirah Abdul HalimlsueyinNo ratings yet
- FullerenesDocument411 pagesFullereneslsueyinNo ratings yet
- Enzyme Kinetics and Catalysis IIDocument36 pagesEnzyme Kinetics and Catalysis IIlsueyinNo ratings yet
- Einstein's Theory of Gravitation: Gravitational WavesDocument9 pagesEinstein's Theory of Gravitation: Gravitational WaveslsueyinNo ratings yet
- Preparation of CatalystsDocument44 pagesPreparation of CatalystslsueyinNo ratings yet
- Potentiometry: Suggested Problems From S.H.C.Document11 pagesPotentiometry: Suggested Problems From S.H.C.lsueyinNo ratings yet
- Persuasive Essay Sample Paper: Name - DateDocument1 pagePersuasive Essay Sample Paper: Name - DatelsueyinNo ratings yet
- Symmetry. Point Groups and Character Tables I, Symmetry Operations and Their Importance For Chemical ProblemsDocument7 pagesSymmetry. Point Groups and Character Tables I, Symmetry Operations and Their Importance For Chemical ProblemslsueyinNo ratings yet
- FTIR IntroductionDocument8 pagesFTIR Introductiontimtailieu2010No ratings yet
- Sample Passing Regents' Essays: 3/2 ModelDocument2 pagesSample Passing Regents' Essays: 3/2 ModellsueyinNo ratings yet
- Chemical Reactions of Copper and Percent YieldDocument8 pagesChemical Reactions of Copper and Percent Yieldlsueyin0% (1)
- Sample Essay Exam ExerciseDocument2 pagesSample Essay Exam ExerciselsueyinNo ratings yet
- Baliga Aviation Light - 183FLPW 1248Document4 pagesBaliga Aviation Light - 183FLPW 1248Sandeep GuptaNo ratings yet
- Amoco ProcessDocument2 pagesAmoco ProcessAddison Juttie100% (3)
- Sainik Class 9 2023 Entrance Exam Question Paper: WWW - Nationaldefenceinstitute.in - +91 9150981461/62/63/64 1Document39 pagesSainik Class 9 2023 Entrance Exam Question Paper: WWW - Nationaldefenceinstitute.in - +91 9150981461/62/63/64 1Kailash KumarNo ratings yet
- Chapter-2-LEVEL MEASUREMENTDocument26 pagesChapter-2-LEVEL MEASUREMENTMohammed YusufNo ratings yet
- Chapter 18 - Fundamentals of Spectrophotometry (Cont'd) AND Chapter 20 - SpectrophotometersDocument18 pagesChapter 18 - Fundamentals of Spectrophotometry (Cont'd) AND Chapter 20 - SpectrophotometersNandhanNo ratings yet
- JR Chemistry Statesofmater emDocument4 pagesJR Chemistry Statesofmater emkrishNo ratings yet
- HVAC System DesignDocument27 pagesHVAC System Designjeanette pao100% (3)
- Sunmica: Basic IngredientsDocument4 pagesSunmica: Basic IngredientssanjuNo ratings yet
- Physical Science q3 m9Document16 pagesPhysical Science q3 m9joanne lanojan100% (2)
- Toward A Standard Thermistor Calibration Method: Data Correction SpreadsheetsDocument7 pagesToward A Standard Thermistor Calibration Method: Data Correction Spreadsheetsjrlr65No ratings yet
- C1152 Cloruro Soluble AcidoDocument4 pagesC1152 Cloruro Soluble Acidoyohanna riveraNo ratings yet
- Chromatographic Methods of AnalysisDocument59 pagesChromatographic Methods of AnalysisCris-Anne Juangco III100% (2)
- Protein Denaturation-Taip VideoDocument2 pagesProtein Denaturation-Taip VideoAmiza HINo ratings yet
- Experiment V: FrictionDocument1 pageExperiment V: FrictionChristopherHiladoNo ratings yet
- Device Technology For Nanoscale III-V Compound Semiconductor Field Effect TransistorsDocument161 pagesDevice Technology For Nanoscale III-V Compound Semiconductor Field Effect TransistorsRaghu Vamsi ChavaliNo ratings yet
- Donald L Smith Gamma PatentDocument34 pagesDonald L Smith Gamma PatentjradNo ratings yet
- Zener PDFDocument3 pagesZener PDFPadmo PadmundonoNo ratings yet
- Old Enthuse Phase 1Document92 pagesOld Enthuse Phase 1bhooshan japeNo ratings yet
- Axial Flow Compressor 2Document11 pagesAxial Flow Compressor 2ram kishor singh100% (1)
- SsssDocument530 pagesSsssasa allahuNo ratings yet
- Bicol College of Applied Science and TechnologyDocument50 pagesBicol College of Applied Science and TechnologyLeo Paulo Del RosarioNo ratings yet
- Chapter 91Document37 pagesChapter 91sscarlett1231No ratings yet
- Powder Dosage FormsDocument3 pagesPowder Dosage FormsMukesh TiwariNo ratings yet
- Confined Space Training: Permit-Required Confined Spaces Title 8 Sections 5156-5159Document31 pagesConfined Space Training: Permit-Required Confined Spaces Title 8 Sections 5156-5159AmeerUlHaqNo ratings yet
- Tension Test ReportDocument7 pagesTension Test ReportTomy GeorgeNo ratings yet
- Concept of Heat TransferDocument27 pagesConcept of Heat Transferdirman dirmanNo ratings yet
- 11 El Chemistry Lesson PlanDocument3 pages11 El Chemistry Lesson PlankrisnuNo ratings yet
- Environment Management Guwahati UniversityDocument17 pagesEnvironment Management Guwahati UniversityRahul DekaNo ratings yet
- Praktikum Bab Karakteristik LED Dan Laser DiodeDocument4 pagesPraktikum Bab Karakteristik LED Dan Laser DiodeErvina IkkeNo ratings yet