You might also like
- Working Guide to Vapor-Liquid Phase Equilibria CalculationsFrom EverandWorking Guide to Vapor-Liquid Phase Equilibria CalculationsRating: 5 out of 5 stars5/5 (1)
- Lecture 26: Non-Ideal Mixtures: 26.1 Recommended TextbooksDocument10 pagesLecture 26: Non-Ideal Mixtures: 26.1 Recommended TextbooksJay SteeleNo ratings yet
- Vapiquid IbriumDocument15 pagesVapiquid IbriumPRAVINNo ratings yet
- Chemical Engineering Thermodynamics Project-I: TopicDocument11 pagesChemical Engineering Thermodynamics Project-I: TopicRohit GuptaNo ratings yet
- Vapor Liquid Equilibria: A Review: Maya B. Mane and S. N. ShindeDocument15 pagesVapor Liquid Equilibria: A Review: Maya B. Mane and S. N. ShindeDesi Riana SaputriNo ratings yet
- Liq Vap Phase 2Document11 pagesLiq Vap Phase 2Xedice MemmedyarovaNo ratings yet
- Vapor Liquid EquilibriumDocument39 pagesVapor Liquid EquilibriumyeenNo ratings yet
- TD BasicsDocument33 pagesTD BasicsKarthik SakthivelNo ratings yet
- Vapour Liquid EquilibriumDocument15 pagesVapour Liquid EquilibriumPRAVINNo ratings yet
- Vle For DummiesDocument8 pagesVle For Dummiesira_rancicNo ratings yet
- Vapor Liquid EquilibriumDocument39 pagesVapor Liquid EquilibriumJakeWilliam100% (1)
- Vap EquilibriumDocument14 pagesVap EquilibriumPRAVINNo ratings yet
- Azeotropic DiagramDocument13 pagesAzeotropic DiagramamoNo ratings yet
- Chapter 6 - Multiphase Systems: CBE2124, LevickyDocument27 pagesChapter 6 - Multiphase Systems: CBE2124, LevickyRimmonNo ratings yet
- Vapor Liquid Equilibria: A Review: ArticleDocument16 pagesVapor Liquid Equilibria: A Review: ArticleLeonardo ReyesNo ratings yet
- 1-Vle Part 1Document30 pages1-Vle Part 1Arfa Zulkifli01No ratings yet
- Vapour Liquid EquilibriumDocument11 pagesVapour Liquid EquilibriumPRAVINNo ratings yet
- Flash CalculationDocument12 pagesFlash CalculationHamidreza HasaniNo ratings yet
- What Is Equilibrium?Document12 pagesWhat Is Equilibrium?PRAVINNo ratings yet
- Chemical Engineering Thermodynamics IIIIIIIIDocument14 pagesChemical Engineering Thermodynamics IIIIIIIIDarnell HendersonNo ratings yet
- Chapter 6. Equilibrium Based Separation Processes: 6.1 Study ObjectivesDocument17 pagesChapter 6. Equilibrium Based Separation Processes: 6.1 Study ObjectivesMaarifa KidogeNo ratings yet
- RS - Lecture 9Document32 pagesRS - Lecture 9Omer IkhlasNo ratings yet
- MomentumDocument20 pagesMomentumMusa MohammedNo ratings yet
- Ec. de Martin PDFDocument17 pagesEc. de Martin PDFMaggyBalcazarNo ratings yet
- Ceic3001 NotesDocument3 pagesCeic3001 NotesZooy2012No ratings yet
- VLE: Vapor Liquid EquilibriumDocument39 pagesVLE: Vapor Liquid EquilibriumTouhid IslamNo ratings yet
- Equilibrium Relationship Between P: A OA ADocument19 pagesEquilibrium Relationship Between P: A OA ATarek ChawafNo ratings yet
- Phase DiagramsDocument19 pagesPhase DiagramsKaaya GodfreyNo ratings yet
- 07-Absorption For HAP and VOCcontrolDocument118 pages07-Absorption For HAP and VOCcontrolTakeshi Tanohuye TanohuyeNo ratings yet
- MomentumDocument20 pagesMomentumMuhammad AdeelNo ratings yet
- On The Demonstration of The Young-Laplace Equation in Introductory Physics CoursesDocument4 pagesOn The Demonstration of The Young-Laplace Equation in Introductory Physics CoursesrobertnwtNo ratings yet
- Jeans Instability and the Criterion for Gravitational CollapseDocument6 pagesJeans Instability and the Criterion for Gravitational CollapseClaudio Cofré MansillaNo ratings yet
- ChE 3G4 Spreadsheet Flash CalculationsDocument9 pagesChE 3G4 Spreadsheet Flash CalculationsRafael Reyes0% (1)
- Forms of The Microscopic Energy Balance: R W T WDocument15 pagesForms of The Microscopic Energy Balance: R W T WTushar BhingradiyaNo ratings yet
- Collocated FVMDocument17 pagesCollocated FVMapoorvs75No ratings yet
- Distillation TheoryDocument40 pagesDistillation TheoryIrvin HernandezNo ratings yet
- Vle 9Document10 pagesVle 9PRAVINNo ratings yet
- Cubic Equations of State-Which Is BestDocument17 pagesCubic Equations of State-Which Is BestvenkieeNo ratings yet
- Chapter 1 - Distillation PDFDocument107 pagesChapter 1 - Distillation PDFFatin Natasha NazriNo ratings yet
- TextDocument4 pagesTextMd Masum BillahNo ratings yet
- CFD MomentumDocument20 pagesCFD MomentumSêlvâkûmâr JayabalaNo ratings yet
- Two-Component Phase Equilibria ExplainedDocument5 pagesTwo-Component Phase Equilibria ExplainedSiti Halimatus Sa'diahNo ratings yet
- Interphase Mass Transfer Theory: y P X PDocument4 pagesInterphase Mass Transfer Theory: y P X PSnape the PrinceNo ratings yet
- FenTrans Parte3Document11 pagesFenTrans Parte3papaboboheNo ratings yet
- Instabilities PDFDocument24 pagesInstabilities PDFCassiano TecchioNo ratings yet
- Fluid Pressure Measurement TechniquesDocument52 pagesFluid Pressure Measurement Techniquesrohit sharma100% (1)
- Single-phase flow porous media pressure equationDocument10 pagesSingle-phase flow porous media pressure equationAnonymous SfJ6jhNo ratings yet
- Review of Phase Equilibria - NEWDocument19 pagesReview of Phase Equilibria - NEWkarmawii taqatqaNo ratings yet
- Vle 4Document15 pagesVle 4PRAVINNo ratings yet
- Unit-1 - FullDocument68 pagesUnit-1 - FullMohammad JunaidNo ratings yet
- 3G4 Flash Calculations1Document9 pages3G4 Flash Calculations1ahmedNo ratings yet
- Introduction to Vapor/Liquid Equilibrium ModelsDocument10 pagesIntroduction to Vapor/Liquid Equilibrium ModelsdawitNo ratings yet
- J.Chem. Ed., 1993, 70 (2), 96Document3 pagesJ.Chem. Ed., 1993, 70 (2), 96IvonneNo ratings yet
- Vapor/Liquid Equilibrium: Mata Kuliah: Termodinamika IIDocument70 pagesVapor/Liquid Equilibrium: Mata Kuliah: Termodinamika IIputri wahyuniNo ratings yet
- Mcelroy 1979Document3 pagesMcelroy 1979t8e7w2koNo ratings yet
- ثرمو2Document19 pagesثرمو2Al-Hassan NeimaNo ratings yet
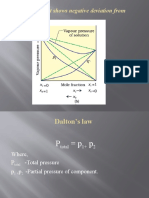
- (B) A Solution That Shows Negative Deviation From Raoult's LawDocument9 pages(B) A Solution That Shows Negative Deviation From Raoult's LawPRAVINNo ratings yet
- The incompressible flow equations: Solving the pressure-velocity couplingDocument10 pagesThe incompressible flow equations: Solving the pressure-velocity couplingnsreddydsceNo ratings yet
- Distillation - Dr.K.Suresh - NotesDocument63 pagesDistillation - Dr.K.Suresh - NotesRoyalNo ratings yet
- Lecture 17: Heat Engines and Possible Processes: 17.1 Recommended TextbooksDocument11 pagesLecture 17: Heat Engines and Possible Processes: 17.1 Recommended TextbooksJay SteeleNo ratings yet
- Lecture 23: Phase Diagrams: 23.1 Recommended TextbooksDocument9 pagesLecture 23: Phase Diagrams: 23.1 Recommended TextbooksJay SteeleNo ratings yet
- Equilibrium and Phase Behavior in Single-Component SystemsDocument11 pagesEquilibrium and Phase Behavior in Single-Component SystemsJay SteeleNo ratings yet
- Fugacity and Departure Functions in Real GasesDocument10 pagesFugacity and Departure Functions in Real GasesJay SteeleNo ratings yet
- Lecture 25: Ideal Mixtures and Raoult's Law: 25.1 Recommended TextbooksDocument10 pagesLecture 25: Ideal Mixtures and Raoult's Law: 25.1 Recommended TextbooksJay SteeleNo ratings yet
- Fugacity: Departure Functions and Single-Component SystemsDocument12 pagesFugacity: Departure Functions and Single-Component SystemsJay SteeleNo ratings yet
- Lecture 16 2019 Thermodynamic Postulates PreclassDocument9 pagesLecture 16 2019 Thermodynamic Postulates PreclassJay SteeleNo ratings yet
- Lecture 16: Postulates of Thermodynamics: 16.1 Recommended TextbooksDocument7 pagesLecture 16: Postulates of Thermodynamics: 16.1 Recommended TextbooksJay SteeleNo ratings yet
- Lecture 22: Equilibrium in Single-Component Systems: 22.1 Recommended TextbooksDocument8 pagesLecture 22: Equilibrium in Single-Component Systems: 22.1 Recommended TextbooksJay SteeleNo ratings yet
- Lecture 25: Ideal Mixtures: 25.1 Recommended TextbooksDocument8 pagesLecture 25: Ideal Mixtures: 25.1 Recommended TextbooksJay SteeleNo ratings yet
- Notation 2019Document3 pagesNotation 2019Jay SteeleNo ratings yet
- Lecture 20: Legendre Transforms: 20.1 Recommended TextbooksDocument10 pagesLecture 20: Legendre Transforms: 20.1 Recommended TextbooksJay SteeleNo ratings yet
- Lecture 19 2019 Fundamental Relation PreclassDocument10 pagesLecture 19 2019 Fundamental Relation PreclassJay SteeleNo ratings yet
- Lecture 21: Conditions of Equilibrium: 21.1 Recommended TextbooksDocument14 pagesLecture 21: Conditions of Equilibrium: 21.1 Recommended TextbooksJay SteeleNo ratings yet
- Efficiency of Reversible Heat EnginesDocument10 pagesEfficiency of Reversible Heat EnginesJay SteeleNo ratings yet
- Fluctuations and Molecular Simulation IntroductionDocument10 pagesFluctuations and Molecular Simulation IntroductionJay SteeleNo ratings yet
- Lecture 15: Free Energy Perturbation: 15.1 Recommended TextbooksDocument6 pagesLecture 15: Free Energy Perturbation: 15.1 Recommended TextbooksJay SteeleNo ratings yet
- Lecture 2 2019 MicrocanonicalDocument9 pagesLecture 2 2019 MicrocanonicalJay SteeleNo ratings yet
- Heat Engine Efficiency and ProcessesDocument7 pagesHeat Engine Efficiency and ProcessesJay SteeleNo ratings yet
- Lecture 18: Entropy: 18.1 Recommended TextbooksDocument8 pagesLecture 18: Entropy: 18.1 Recommended TextbooksJay SteeleNo ratings yet
- Lecture 20: Legendre Transforms: 20.1 Recommended TextbooksDocument10 pagesLecture 20: Legendre Transforms: 20.1 Recommended TextbooksJay SteeleNo ratings yet
- Lecture 13: The Structure of Classical Fluids: 13.1 Recommended TextbooksDocument10 pagesLecture 13: The Structure of Classical Fluids: 13.1 Recommended TextbooksJay SteeleNo ratings yet
- Lecture 8: The Ising Model and Mean-Field Theory: 8.1 Recommended Textbook Chapters For This SectionDocument7 pagesLecture 8: The Ising Model and Mean-Field Theory: 8.1 Recommended Textbook Chapters For This SectionJay SteeleNo ratings yet
- Generalized EnsemblesDocument9 pagesGeneralized EnsemblesJay SteeleNo ratings yet
- Monte Carlo Simulations ExplainedDocument12 pagesMonte Carlo Simulations ExplainedJay SteeleNo ratings yet
- Lecture 15: Free Energy Perturbation: 15.1 Recommended TextbooksDocument8 pagesLecture 15: Free Energy Perturbation: 15.1 Recommended TextbooksJay SteeleNo ratings yet
- Canonical Partition Function DerivationDocument9 pagesCanonical Partition Function DerivationJay SteeleNo ratings yet
- Lecture 12: MD Thermostats: 12.1 Recommended TextbooksDocument10 pagesLecture 12: MD Thermostats: 12.1 Recommended TextbooksJay SteeleNo ratings yet
- QualitativeDocument306 pagesQualitativeParintins Parintins100% (1)
- Drilling Soal TBI 2Document6 pagesDrilling Soal TBI 2jendeuk kimNo ratings yet
- The Nature of Emotion Fundamental QuestiDocument4 pagesThe Nature of Emotion Fundamental Questimarllon jonasNo ratings yet
- Eddo 2020Document65 pagesEddo 2020temesgenNo ratings yet
- Melanie Donato Loyola Seeks Agricultural Technologist RoleDocument5 pagesMelanie Donato Loyola Seeks Agricultural Technologist RoleIan Dante ArcangelesNo ratings yet
- Prasetyawan 2020Document9 pagesPrasetyawan 2020Tania CanchanyaNo ratings yet
- Pianti Pelajaran KenuanDocument93 pagesPianti Pelajaran KenuanUziNo ratings yet
- Conditional Clause Types 2 - BayBeeDocument3 pagesConditional Clause Types 2 - BayBeecrrrNo ratings yet
- Differential EquationsDocument4 pagesDifferential EquationstuaNo ratings yet
- Answers Unit 5Document2 pagesAnswers Unit 5FranciscaBalasSuarezNo ratings yet
- Reading Passage 1: Swallows in MigrationDocument3 pagesReading Passage 1: Swallows in Migrationhtrang deyyNo ratings yet
- Akta Pure Chromatography SystemDocument14 pagesAkta Pure Chromatography SystemCeli OlmedoNo ratings yet
- Steel Forgings, Carbon and Alloy, For General Industrial UseDocument9 pagesSteel Forgings, Carbon and Alloy, For General Industrial UseHugo SiqueiraNo ratings yet
- Black Widow Medley Throughout The MCU Various ComposersDocument6 pagesBlack Widow Medley Throughout The MCU Various ComposersPiano EasyNo ratings yet
- D.5 Test Yourself QuestionsDocument2 pagesD.5 Test Yourself QuestionsTaeha KimNo ratings yet
- Understanding Social WorkDocument7 pagesUnderstanding Social WorkIsis Angelica FarinasNo ratings yet
- s4 Reading VocabularyDocument45 pagess4 Reading VocabularyMd Towhidul IslamNo ratings yet
- Architecture Presentation Board TipsDocument16 pagesArchitecture Presentation Board TipsGeni Rapsanjani100% (1)
- Electronic Circuit Style CV by SlidesgoDocument39 pagesElectronic Circuit Style CV by SlidesgoHelmanz NugrahaNo ratings yet
- State-of-the-Art For Small Satellite Propulsion Systems: Code 597.0 - Propulsion Branch NASA/Goddard Space Flight CenterDocument8 pagesState-of-the-Art For Small Satellite Propulsion Systems: Code 597.0 - Propulsion Branch NASA/Goddard Space Flight CenterMauricio JandirNo ratings yet
- Week 2 ELDocument17 pagesWeek 2 ELSyryll FloresNo ratings yet
- SONDEX TRAINING MANUAL TWO PHASE PRODUCTION LOG ANALYSISDocument1 pageSONDEX TRAINING MANUAL TWO PHASE PRODUCTION LOG ANALYSISKader BakourNo ratings yet
- STE NotesDocument4 pagesSTE NotesDana VillanoNo ratings yet
- Botanical Drawing by Walter Hood Fitch (1817-1892)Document13 pagesBotanical Drawing by Walter Hood Fitch (1817-1892)Monica Perez100% (1)
- Coffey Mining MapInfo Advanced 2009Document58 pagesCoffey Mining MapInfo Advanced 2009Ahmed GhoneimNo ratings yet
- IJLL - SanskritDocument6 pagesIJLL - SanskritSudarshan RaghununanNo ratings yet
- Class 2 NSO Preparation Free Sample Test: Aabbaa Ababab Abbabb Aababb A. B. C. DDocument11 pagesClass 2 NSO Preparation Free Sample Test: Aabbaa Ababab Abbabb Aababb A. B. C. DamarpreetsethiNo ratings yet
- Bronze Age EuropeDocument10 pagesBronze Age EuropeVíctor Alfonso Medina LugoNo ratings yet
- Energy Fundamental System Analysis Demand in National Capital RegionDocument14 pagesEnergy Fundamental System Analysis Demand in National Capital RegionMaher Marquez100% (1)
- Cell TheoryDocument31 pagesCell TheoryMarc Ian Young100% (1)