You might also like
- Practical Handbook of Pharmaceutical Chemistry for M.PharmFrom EverandPractical Handbook of Pharmaceutical Chemistry for M.PharmNo ratings yet
- Determination of Guava (Psidium Guajava L.) LeafDocument13 pagesDetermination of Guava (Psidium Guajava L.) LeafTiara Nugraeni EkaNo ratings yet
- FTB 51 1 084 091Document8 pagesFTB 51 1 084 091erni yunitaNo ratings yet
- HPLC-DAD-ESI-MS Analysis of Phenolic Compounds in Tomato FruitDocument6 pagesHPLC-DAD-ESI-MS Analysis of Phenolic Compounds in Tomato Fruitnatale_carvalhoNo ratings yet
- 06 Ac17 PDFDocument17 pages06 Ac17 PDFMuhammad Abdur RokhimNo ratings yet
- Strassmann, 2008Document6 pagesStrassmann, 2008Natália AguiarNo ratings yet
- Structure Antioxidant Efficiency Relationships of Phenolic Compounds and Their Contribution To The Antioxidant Activity of Sea Buckthorn JuiceDocument7 pagesStructure Antioxidant Efficiency Relationships of Phenolic Compounds and Their Contribution To The Antioxidant Activity of Sea Buckthorn JuicejohnheverthNo ratings yet
- Pharmacochemical Study of Aqueous Extracts of Passiflora Alata Dryander and Passiflora Edulis SimsDocument4 pagesPharmacochemical Study of Aqueous Extracts of Passiflora Alata Dryander and Passiflora Edulis SimsGabrielNo ratings yet
- Phenolic Composition and Antioxidant Properties of Some Traditionally Used Medicinal Plants Affected by The Extraction Time and HydrolysisDocument9 pagesPhenolic Composition and Antioxidant Properties of Some Traditionally Used Medicinal Plants Affected by The Extraction Time and HydrolysisBeatriz MelloNo ratings yet
- Food Chemistry: Haejin Bae, G.K. Jayaprakasha, John Jifon, Bhimanagouda S. PatilDocument8 pagesFood Chemistry: Haejin Bae, G.K. Jayaprakasha, John Jifon, Bhimanagouda S. Patilreza rezaieNo ratings yet
- Mcf. Articulo1posgrado 2023-2sDocument7 pagesMcf. Articulo1posgrado 2023-2sHector Santiago Lopez AcostaNo ratings yet
- Dandelion Phenolic Compounds..Document22 pagesDandelion Phenolic Compounds..gail CieloNo ratings yet
- 1 s2.0 S0102695X15000332 MainDocument6 pages1 s2.0 S0102695X15000332 MainMian KashifNo ratings yet
- 43 83 1 SMDocument6 pages43 83 1 SMyulinarNo ratings yet
- Microwave Extraction of Flavonoids from Phyllanthus emblicaDocument15 pagesMicrowave Extraction of Flavonoids from Phyllanthus emblicaDr. Yedhu Krishnan RNo ratings yet
- Toxicity EvaluationDocument12 pagesToxicity EvaluationEduSmart HubNo ratings yet
- Partial Characterization and Anticoagulant Activity of A Heterofucan From The Brown Seaweed Padina GymnosporaDocument11 pagesPartial Characterization and Anticoagulant Activity of A Heterofucan From The Brown Seaweed Padina Gymnosporaanon_743258777No ratings yet
- Isolation of Flavonoid from Launaea procumbens by HPTLCDocument7 pagesIsolation of Flavonoid from Launaea procumbens by HPTLCbledok khatolNo ratings yet
- Chen12-Flavonoid-Shift ReagentsDocument26 pagesChen12-Flavonoid-Shift ReagentsMarvinNo ratings yet
- 5c0a PDFDocument9 pages5c0a PDFnelisaNo ratings yet
- Za2 PDFDocument7 pagesZa2 PDFChristine Yohana SianturiNo ratings yet
- Aloe VeraDocument0 pagesAloe VeraSyahrul FarhanahNo ratings yet
- Flavonoids in Juglans Regia L. Leaves and Evaluation of in Vitro Antioxidant Activity Via Intracellular and Chemical MethodsDocument10 pagesFlavonoids in Juglans Regia L. Leaves and Evaluation of in Vitro Antioxidant Activity Via Intracellular and Chemical MethodsDyanne NegruNo ratings yet
- Analysis of Antioxidative Phenolic Compounds in ArtichokeDocument8 pagesAnalysis of Antioxidative Phenolic Compounds in ArtichokeAhmet UluadNo ratings yet
- Kirinyuh LengkpDocument22 pagesKirinyuh LengkpPutrii SerlindaNo ratings yet
- TammaDocument7 pagesTammaنورالدين غرافNo ratings yet
- Giuffrida Et Al-2012-Phytochemical AnalysisDocument7 pagesGiuffrida Et Al-2012-Phytochemical AnalysisNatalia NetrebaNo ratings yet
- Research Article: UPLC-DAD-QTOF/MS Analysis of Flavonoids From 12 Varieties of Korean Mulberry FruitDocument9 pagesResearch Article: UPLC-DAD-QTOF/MS Analysis of Flavonoids From 12 Varieties of Korean Mulberry Fruittayyaba mehmoodNo ratings yet
- Kuersetin With HCLDocument5 pagesKuersetin With HCLavivahNo ratings yet
- DescargaDocument20 pagesDescargaKarla Argelia Petit ArteagaNo ratings yet
- Jurnal PenelitianDocument7 pagesJurnal PenelitianVera FebriantiNo ratings yet
- Bioassay-Guided Isolation of Flavonoids From Caesalpinia Bonduc (L.) Roxb. and Evaluation of Their CytotoxicityDocument9 pagesBioassay-Guided Isolation of Flavonoids From Caesalpinia Bonduc (L.) Roxb. and Evaluation of Their Cytotoxicitynurul hidayatiNo ratings yet
- Shipra Annona PaperDocument5 pagesShipra Annona PaperMuhammad Fattah FazelNo ratings yet
- Arquivo ABrabDocument7 pagesArquivo ABrabWeslei Bruno BoteroNo ratings yet
- Research Article Eugenia Uniflora L. (Myrtaceae) Leaves and Its Ultrasound-AssistedDocument10 pagesResearch Article Eugenia Uniflora L. (Myrtaceae) Leaves and Its Ultrasound-AssistedKELLY JOHANA PEDROZA BERRIONo ratings yet
- CHEMICAL CONSTITUENTS FROM VITIS VINIFERA LEAVES AND BRANCHESDocument4 pagesCHEMICAL CONSTITUENTS FROM VITIS VINIFERA LEAVES AND BRANCHESSandra Marcela PabónNo ratings yet
- 09e41508ace8b77161000000 PDFDocument7 pages09e41508ace8b77161000000 PDFNandika Kasaru JackNo ratings yet
- Jurnal Saponin PDFDocument5 pagesJurnal Saponin PDFAnisa Uswatun KhasanahNo ratings yet
- Enginar Atığı Fenolik MaddeleriDocument16 pagesEnginar Atığı Fenolik MaddeleriAhmet UluadNo ratings yet
- 6cnYgB 1Document5 pages6cnYgB 1Moses FernandoNo ratings yet
- 05 Intan Soraya Che Sulaiman - Paling FunctionDocument14 pages05 Intan Soraya Che Sulaiman - Paling FunctionIdham ZaharudieNo ratings yet
- Analysis of Selected Phenolic Acids and Flavonoids in Amaranthus Cruentus and by HPLCDocument12 pagesAnalysis of Selected Phenolic Acids and Flavonoids in Amaranthus Cruentus and by HPLCWillam PariNo ratings yet
- Cardaria DrabaDocument4 pagesCardaria DrabaMangiNo ratings yet
- Phytochemical Studies of Medicinal Plants From Several Genera With Antidiabetic ActivitiesDocument25 pagesPhytochemical Studies of Medicinal Plants From Several Genera With Antidiabetic ActivitiesPrograma BRICNo ratings yet
- Qualitative and Quantitative Phytochemical Evaluations of Strophanthus Hispidus Stem BarkDocument5 pagesQualitative and Quantitative Phytochemical Evaluations of Strophanthus Hispidus Stem BarkInternational Organization of Scientific Research (IOSR)No ratings yet
- J Foodchem 2004 01 055Document7 pagesJ Foodchem 2004 01 055Rajan PandaNo ratings yet
- Article Final Warionia Flavonoids ChamcDocument12 pagesArticle Final Warionia Flavonoids ChamcMed AjNo ratings yet
- 2015 3 2 10 AlebiosuDocument7 pages2015 3 2 10 AlebiosudanilriosNo ratings yet
- Isolation of Flavonoid Constituent From Launaea Procumbens Roxb. by Preparative HPTLC MethodDocument7 pagesIsolation of Flavonoid Constituent From Launaea Procumbens Roxb. by Preparative HPTLC MethodIOSR Journal of PharmacyNo ratings yet
- Jurnal 5Document5 pagesJurnal 5Khafiya KimNo ratings yet
- 10 - Persic - Short CommunicationDocument6 pages10 - Persic - Short Communicationsabrinechelly09No ratings yet
- Application of A UV-vis detection-HPLC Method For ADocument9 pagesApplication of A UV-vis detection-HPLC Method For Alili&valiNo ratings yet
- Extraction of Phenolic Compounds From Blueberry (Vaccinium Myrtillus L.) RESIDUES USING SUPERCRITICAL CO and Pressurized WaterDocument10 pagesExtraction of Phenolic Compounds From Blueberry (Vaccinium Myrtillus L.) RESIDUES USING SUPERCRITICAL CO and Pressurized WaterKAROL ELIZABETH OBANDO AMAYANo ratings yet
- Legall 2003Document9 pagesLegall 2003nguyễn thủyNo ratings yet
- Tugas Jurnal DNW - 21.3.23 PDFDocument6 pagesTugas Jurnal DNW - 21.3.23 PDFsalsabila JacobNo ratings yet
- 10.1007@s40005 020 00477 WDocument16 pages10.1007@s40005 020 00477 WVj McNo ratings yet
- Anita Wagh-39-Vol.-10-Issue-6-June-2019-IJPSR-RA-11001 PDFDocument6 pagesAnita Wagh-39-Vol.-10-Issue-6-June-2019-IJPSR-RA-11001 PDFbutlesrNo ratings yet
- Shen, Y. (2016) - Butterfly Pea (Clitoria Ternatea) Seed and Petal Extracts Decreased HEp-2 Carcinoma Cell ViabilityDocument9 pagesShen, Y. (2016) - Butterfly Pea (Clitoria Ternatea) Seed and Petal Extracts Decreased HEp-2 Carcinoma Cell ViabilityMauricio AvilaNo ratings yet
- Environmental Metabolomics: Applications in field and laboratory studies to understand from exposome to metabolomeFrom EverandEnvironmental Metabolomics: Applications in field and laboratory studies to understand from exposome to metabolomeDiana Alvarez-MunozNo ratings yet
- Aroma Characterisation of Gouda-Type CheesesDocument11 pagesAroma Characterisation of Gouda-Type CheesescsandrasNo ratings yet
- Accumulation of Short N-Chain Ethyl Esters by Esterases of Lactic Acid Bacteria Under Conditions Simulating Ripening Parmesan CheeseDocument8 pagesAccumulation of Short N-Chain Ethyl Esters by Esterases of Lactic Acid Bacteria Under Conditions Simulating Ripening Parmesan CheesecsandrasNo ratings yet
- The Repellent and Persistent Toxic Effects of Essential Oils Against The Poultry Red Mite, Dermanyssus GallinaeDocument5 pagesThe Repellent and Persistent Toxic Effects of Essential Oils Against The Poultry Red Mite, Dermanyssus GallinaecsandrasNo ratings yet
- Flavour Formation by Lactic Acid Bacteria and Biochemical Flavour Profiling of Cheese ProductsDocument20 pagesFlavour Formation by Lactic Acid Bacteria and Biochemical Flavour Profiling of Cheese ProductscsandrasNo ratings yet
- WineCheeseCracker PairingGuide PDFDocument1 pageWineCheeseCracker PairingGuide PDFCza SurlaNo ratings yet
- Changes in The Volatile Composition of A Semihard Ewe Milk Cheese Induced by High-Pressure Treatment of 300 MPaDocument8 pagesChanges in The Volatile Composition of A Semihard Ewe Milk Cheese Induced by High-Pressure Treatment of 300 MPacsandrasNo ratings yet
- Evaluation of Ideal Wine and Cheese Pairs Using A Deviation-From-Ideal Scale With Food and Wine ExpertsDocument13 pagesEvaluation of Ideal Wine and Cheese Pairs Using A Deviation-From-Ideal Scale With Food and Wine ExpertscsandrasNo ratings yet
- Environmental Interactions With The Toxicity of Plant Essential Oils To The Poultry Red Mite Dermanyssus GallinaeDocument8 pagesEnvironmental Interactions With The Toxicity of Plant Essential Oils To The Poultry Red Mite Dermanyssus GallinaecsandrasNo ratings yet
- Acaricidal and Repellent Effects of Essential Oils Against Ticks A ReviewDocument17 pagesAcaricidal and Repellent Effects of Essential Oils Against Ticks A ReviewcsandrasNo ratings yet
- Carvacrol As A Potent Natural Acaricide Against Dermanyssus GallinaeDocument6 pagesCarvacrol As A Potent Natural Acaricide Against Dermanyssus GallinaecsandrasNo ratings yet
- Laboratory Tests For Controlling Poultry Red Mites (Dermanyssus Gallinae) With Predatory Mites in Small Laying Hen' CagesDocument13 pagesLaboratory Tests For Controlling Poultry Red Mites (Dermanyssus Gallinae) With Predatory Mites in Small Laying Hen' CagescsandrasNo ratings yet
- Food and Chemical Toxicology: SciencedirectDocument7 pagesFood and Chemical Toxicology: SciencedirectcsandrasNo ratings yet
- Should The Poultry Red Mite Dermanyssus Gallinae Be of Wider Concern For Veterinary and Medical ScienceDocument10 pagesShould The Poultry Red Mite Dermanyssus Gallinae Be of Wider Concern For Veterinary and Medical SciencecsandrasNo ratings yet
- Active Components of Essential Oils As Acaricides Against Dermanyssus GallinaeDocument15 pagesActive Components of Essential Oils As Acaricides Against Dermanyssus GallinaecsandrasNo ratings yet
- Mode of Action and Variability in Efficacy of Plant Essential Oils Showing Toxicity Against The Poultry Red Mite, Dermanyssus GallinaeDocument7 pagesMode of Action and Variability in Efficacy of Plant Essential Oils Showing Toxicity Against The Poultry Red Mite, Dermanyssus GallinaecsandrasNo ratings yet
- The Effect of Insecticide Synergists On The Response of Scabies Mites To Pyrethroid AcaricidesDocument8 pagesThe Effect of Insecticide Synergists On The Response of Scabies Mites To Pyrethroid AcaricidescsandrasNo ratings yet
- Toxicity of Plant Essential Oils To Different Life Stages of The Poultry Red Mite, Dermanyssus Gallinae, and Non-Target InvertebratesDocument7 pagesToxicity of Plant Essential Oils To Different Life Stages of The Poultry Red Mite, Dermanyssus Gallinae, and Non-Target InvertebratescsandrasNo ratings yet
- Plant Essential Oils as Potential Dermanyssus gallinae RepellentsDocument6 pagesPlant Essential Oils as Potential Dermanyssus gallinae RepellentscsandrasNo ratings yet
- Lack of Prolonged Activity of Lavender Essential Oils As Acaricides Against The Poultry Red Mite (Dermanyssusgallinae) Under Laboratory ConditionsDocument3 pagesLack of Prolonged Activity of Lavender Essential Oils As Acaricides Against The Poultry Red Mite (Dermanyssusgallinae) Under Laboratory ConditionscsandrasNo ratings yet
- Acaricidal Activity of Eugenol Based Compounds Against Scabies MiteDocument9 pagesAcaricidal Activity of Eugenol Based Compounds Against Scabies MitecsandrasNo ratings yet
- Veterinary Parasitology: D.R. George, G. Olatunji, J.H. Guy, O.A.E. SparaganoDocument4 pagesVeterinary Parasitology: D.R. George, G. Olatunji, J.H. Guy, O.A.E. SparaganocsandrasNo ratings yet
- Background To New Treatments For COVID-19, Including Its Chronicity, Through Altering Elements of The Cytokine StormDocument13 pagesBackground To New Treatments For COVID-19, Including Its Chronicity, Through Altering Elements of The Cytokine StormcsandrasNo ratings yet
- A Hypothesized Role For Dysregulated Bradykinin Signaling in COVID-19 Respiratory ComplicationsDocument5 pagesA Hypothesized Role For Dysregulated Bradykinin Signaling in COVID-19 Respiratory ComplicationscsandrasNo ratings yet
- SARS-CoV-2 Suppresses Anticoagulant and Fibrinolytic Gene Expression in The LungDocument13 pagesSARS-CoV-2 Suppresses Anticoagulant and Fibrinolytic Gene Expression in The LungcsandrasNo ratings yet
- Journal of Functional Foods: SciencedirectDocument8 pagesJournal of Functional Foods: SciencedirectDelfina RubioNo ratings yet
- Altered Smell and Taste Anosmia, Parosmia and The Impact of Long Covid-19Document18 pagesAltered Smell and Taste Anosmia, Parosmia and The Impact of Long Covid-19csandrasNo ratings yet
- Acaricidal Effect of Eleven Essential Oils Against The Poultry Red Mite Dermanyssus Gallinae (Acari Dermanyssidae)Document6 pagesAcaricidal Effect of Eleven Essential Oils Against The Poultry Red Mite Dermanyssus Gallinae (Acari Dermanyssidae)csandrasNo ratings yet
- Individuals Cannot Rely On COVID-19 Herd Immunity Durable Immunity To Viral Disease Is Limited To Viruses With Obligate Viremic SpreadDocument4 pagesIndividuals Cannot Rely On COVID-19 Herd Immunity Durable Immunity To Viral Disease Is Limited To Viruses With Obligate Viremic SpreadcsandrasNo ratings yet
- Prevalence and Persistence of Smell and Taste Dysfunction in COVID-19Document2 pagesPrevalence and Persistence of Smell and Taste Dysfunction in COVID-19csandrasNo ratings yet
- Individuals Cannot Rely On COVID-19 Herd Immunity Durable Immunity To Viral Disease Is Limited To Viruses With Obligate Viremic SpreadDocument4 pagesIndividuals Cannot Rely On COVID-19 Herd Immunity Durable Immunity To Viral Disease Is Limited To Viruses With Obligate Viremic SpreadcsandrasNo ratings yet
- TB Diode 3211430Document27 pagesTB Diode 3211430adamnorazlanNo ratings yet
- Flexural Strength of Ferrocement PannelDocument51 pagesFlexural Strength of Ferrocement PannelAishwarya ShedgeNo ratings yet
- HBSE 2019 12th ChemistryDocument64 pagesHBSE 2019 12th ChemistrytempraryNo ratings yet
- 9701 Scheme of Work (For Examination From 2022) - 3Document1 page9701 Scheme of Work (For Examination From 2022) - 3DuckyNo ratings yet
- Resistivity and Resistance CalculationsDocument5 pagesResistivity and Resistance CalculationsJhac FamorNo ratings yet
- Jarissa Banner Nitration of Bromobnzene LabDocument15 pagesJarissa Banner Nitration of Bromobnzene LabJuiloNo ratings yet
- Chemistry: NEET - 2020-21Document1 pageChemistry: NEET - 2020-21pratikNo ratings yet
- CustomBiotech Cedex IgG AssayDocument4 pagesCustomBiotech Cedex IgG Assayaamin farasNo ratings yet
- Industrial ElectrochemDocument11 pagesIndustrial ElectrochemHassan AgNo ratings yet
- Mdcp21% TdsDocument2 pagesMdcp21% TdsOSCAR ADOLFO HERNANDEZ PIRIRNo ratings yet
- GRP Lamination Processing - Instructions PDFDocument34 pagesGRP Lamination Processing - Instructions PDFmuhammadkashikNo ratings yet
- Nova Prime & Nova Plus ABG Analyzer BrochureDocument2 pagesNova Prime & Nova Plus ABG Analyzer BrochureMuhammad BilalNo ratings yet
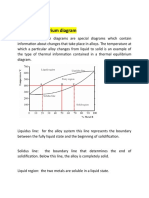
- Task 4: Thermal Equilibrium DiagramDocument3 pagesTask 4: Thermal Equilibrium DiagramMisbah Tehseen100% (1)
- Rahul International School's PaperDocument8 pagesRahul International School's PaperHarshal MadaviNo ratings yet
- Laser GraftingDocument5 pagesLaser GraftingpcnferreiraNo ratings yet
- Exam SolutDocument12 pagesExam Solutnunulovin911No ratings yet
- The Mystery of Molten Metal: Natalia Sobczak, Jerzy Sobczak, Rajiv Asthana and Robert PurgertDocument13 pagesThe Mystery of Molten Metal: Natalia Sobczak, Jerzy Sobczak, Rajiv Asthana and Robert Purgertyosua l.rajaNo ratings yet
- Keep That Mimosa: Mimosa Tenuiflora ( M. Hostilis) Root-BarkDocument5 pagesKeep That Mimosa: Mimosa Tenuiflora ( M. Hostilis) Root-BarkjajcabNo ratings yet
- Solubility Isotherms Sulfate-Ethylene Glycol-Water: SodiumDocument6 pagesSolubility Isotherms Sulfate-Ethylene Glycol-Water: SodiumSagarNo ratings yet
- Comparative Analysis of Smokeless Gunpowders by Fourier Transform Infrared and Raman SpectrosDocument8 pagesComparative Analysis of Smokeless Gunpowders by Fourier Transform Infrared and Raman SpectrosmrenaultNo ratings yet
- Immobilization of E. Coli Expressing Bacillus Pumilus CynD in Three Organic Polymer MatricesDocument23 pagesImmobilization of E. Coli Expressing Bacillus Pumilus CynD in Three Organic Polymer MatricesLUIS CARLOS ROMERO ZAPATANo ratings yet
- 2023-MQP-12334 CHEMISTRYrfDocument6 pages2023-MQP-12334 CHEMISTRYrfVenu gopal PreethamNo ratings yet
- Additives, Driers, Accelerators and Catalysts: For Coatings, Paints, Composites, Printing Inks and AdhesivesDocument16 pagesAdditives, Driers, Accelerators and Catalysts: For Coatings, Paints, Composites, Printing Inks and AdhesivesAnanthanarayananNo ratings yet
- Experimental Study of The Physical Properties of Concrete Prepared by Partial Replacement of Cement With Alccofine, Metakaolite and GGBSDocument21 pagesExperimental Study of The Physical Properties of Concrete Prepared by Partial Replacement of Cement With Alccofine, Metakaolite and GGBSIJRASETPublicationsNo ratings yet
- Footing Design (WSD Method) PDFDocument2 pagesFooting Design (WSD Method) PDFFaruque AbdullahNo ratings yet
- An Fat Soluble Carotenoid infinityII 6470a Poroshell 5994 5064en AgilentDocument11 pagesAn Fat Soluble Carotenoid infinityII 6470a Poroshell 5994 5064en AgilentLaura Tatiana AguirreNo ratings yet
- Characterization, Beneficiation and Utilization of A Kaolinite ClayDocument11 pagesCharacterization, Beneficiation and Utilization of A Kaolinite ClayArun Ebenezer100% (1)
- BS en 01905-1999Document8 pagesBS en 01905-1999mohamed guedichiNo ratings yet
- AIATS Schedule for Class XIIDocument5 pagesAIATS Schedule for Class XIIPARSHV MESHIYANo ratings yet
- CHM361 - CHAPTER 2 Molecular Orbital TheoryDocument35 pagesCHM361 - CHAPTER 2 Molecular Orbital TheoryEhaz100% (1)