You might also like
- Astrology Chart Key PDFDocument1 pageAstrology Chart Key PDFganapathi_gNo ratings yet
- The Dynamics of Tibetan Singing BowlsDocument22 pagesThe Dynamics of Tibetan Singing BowlsmepurquaNo ratings yet
- Body Bulder Guide New GIGADocument272 pagesBody Bulder Guide New GIGAMuhammad Hanif FuadiNo ratings yet
- The Cinematic VR Field GuideDocument68 pagesThe Cinematic VR Field GuideGrant Anderson100% (1)
- Xerox ColorQube 9303 Family Service ManualDocument1,812 pagesXerox ColorQube 9303 Family Service ManualДмитрий Разуваев100% (1)
- FCC Process Fundamentals & Technology EvolutionDocument42 pagesFCC Process Fundamentals & Technology Evolutionranjith_asp0% (1)
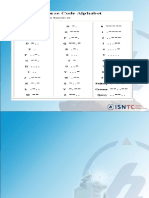
- (A) Morse Code and International Code of Flag SignalsDocument63 pages(A) Morse Code and International Code of Flag SignalsJohn Rey CastillanoNo ratings yet
- Dasa Bukthi Anthara Pala PDFDocument18 pagesDasa Bukthi Anthara Pala PDFsarathNo ratings yet
- Aqap 2105 2019 Eng DataDocument26 pagesAqap 2105 2019 Eng Data신동득No ratings yet
- Textile Processing NotesDocument15 pagesTextile Processing NotespithadiyatrushaNo ratings yet
- RCC93 Flat Slabs (Tables)Document24 pagesRCC93 Flat Slabs (Tables)Gan Chin PhangNo ratings yet
- Aguennouz Et Al 2016Document9 pagesAguennouz Et Al 2016Dário Talita SantanaNo ratings yet
- The Genetic Aspect and Morphological Appearance of AchondrogenesisDocument10 pagesThe Genetic Aspect and Morphological Appearance of AchondrogenesisRizka RamadaniNo ratings yet
- Bookshelf NBK11546Document24 pagesBookshelf NBK11546Brew-sam ABNo ratings yet
- A Glycolytic Shift in Schwann Cells Supports Injured Axons-1Document1 pageA Glycolytic Shift in Schwann Cells Supports Injured Axons-1api-655391815No ratings yet
- tmp5696 TMPDocument10 pagestmp5696 TMPFrontiersNo ratings yet
- Deficient Autophagy in Microglia Impairs Synaptic PruningDocument9 pagesDeficient Autophagy in Microglia Impairs Synaptic Pruningravenclaw valdezNo ratings yet
- Sid-5 Is An Endosome-Associated Protein Required For Efficient Systemic Rnai in C. ElegansDocument6 pagesSid-5 Is An Endosome-Associated Protein Required For Efficient Systemic Rnai in C. EleganskaeterineNo ratings yet
- Immunoglobulin ADocument11 pagesImmunoglobulin AFIAL FAIDA ASSYIFANo ratings yet
- tmp37AA TMPDocument12 pagestmp37AA TMPFrontiersNo ratings yet
- ADAMTS-1 Cleaves A Cartilage Proteoglycan, Aggrecan 00Document5 pagesADAMTS-1 Cleaves A Cartilage Proteoglycan, Aggrecan 00Sonia Barbosa CornelioNo ratings yet
- E2f Nature 2009Document5 pagesE2f Nature 2009Danian ChenNo ratings yet
- TMP DF16Document6 pagesTMP DF16FrontiersNo ratings yet
- RT Paper For PresentationDocument27 pagesRT Paper For PresentationYinebeb MezgebuNo ratings yet
- Magnesium Ion Influx Reduces Neuroinflammation in Ab Precursor Protein/presenilin 1 Transgenic Mice by Suppressing The Expression of Interleukin-1bDocument14 pagesMagnesium Ion Influx Reduces Neuroinflammation in Ab Precursor Protein/presenilin 1 Transgenic Mice by Suppressing The Expression of Interleukin-1bkleberttoscanoNo ratings yet
- Seminario 3 - Role of Dystrophin and Utrophin For Assembly and FunctionDocument18 pagesSeminario 3 - Role of Dystrophin and Utrophin For Assembly and Functiondmalaga06No ratings yet
- Michael A. Dyer and Constance L. Cepko - Control of Müller Glial Cell Proliferation and Activation Following Retinal InjuryDocument8 pagesMichael A. Dyer and Constance L. Cepko - Control of Müller Glial Cell Proliferation and Activation Following Retinal InjuryHutsDMNo ratings yet
- Ferrara 2003Document8 pagesFerrara 2003Chaima DjabouNo ratings yet
- Altered Neurogenesis in Mouse Models of Alzheimer DiseaseDocument10 pagesAltered Neurogenesis in Mouse Models of Alzheimer Diseaseapi-682804894No ratings yet
- BMP-2 and CDMP-2: Stimulation of Chondrocyte Production of ProteoglycanDocument8 pagesBMP-2 and CDMP-2: Stimulation of Chondrocyte Production of ProteoglycanFrank ZhouNo ratings yet
- Aberrant Upregulation of The Glycolytic Enzyme PFKFB3 in CLN7 Neuronal Ceroid LipofuscinosisDocument14 pagesAberrant Upregulation of The Glycolytic Enzyme PFKFB3 in CLN7 Neuronal Ceroid Lipofuscinosisezequiel.grondona.cbaNo ratings yet
- Alpha-Gal Detectors in Xenotransplantation Research: A Word of CautionDocument6 pagesAlpha-Gal Detectors in Xenotransplantation Research: A Word of CautionAnonymous iqoU1mtNo ratings yet
- Evolution Need PeriodDocument6 pagesEvolution Need PeriodRaviNo ratings yet
- Glutamate Excitotoxicity in The Cerebellum Mediated by IL-1: Journal ClubDocument3 pagesGlutamate Excitotoxicity in The Cerebellum Mediated by IL-1: Journal ClubJar JarNo ratings yet
- Muscular DystrophyDocument10 pagesMuscular DystrophyPradhnesh AndhareNo ratings yet
- (論文閱讀) 通過基因編輯確認Nlgn3基因與自閉症有關Document24 pages(論文閱讀) 通過基因編輯確認Nlgn3基因與自閉症有關徐No ratings yet
- Celsr3 Is Required For Normal Development of GABA Circuits in The Inner RetinaDocument12 pagesCelsr3 Is Required For Normal Development of GABA Circuits in The Inner RetinamacadamiannutNo ratings yet
- PpokDocument7 pagesPpokSkoy KuyNo ratings yet
- Intersectin Regulates Dendritic Spine Development and Somatodendritic Endocytosis But Not Synaptic Vesicle Recycling in Hippocampal NeuronsDocument10 pagesIntersectin Regulates Dendritic Spine Development and Somatodendritic Endocytosis But Not Synaptic Vesicle Recycling in Hippocampal NeuronsSergeat18BNo ratings yet
- Jenniskens 2006 Biochem + Funct Modul Coll Netwerk TGFB EtcDocument11 pagesJenniskens 2006 Biochem + Funct Modul Coll Netwerk TGFB EtcH JahrNo ratings yet
- Orphan Receptor GPR158 Serves As A Metabotropic Glycine Receptor mGlyRDocument7 pagesOrphan Receptor GPR158 Serves As A Metabotropic Glycine Receptor mGlyRStress LessNo ratings yet
- Estrada FS Et Al Glial Activation in Pilocarpine 2012Document6 pagesEstrada FS Et Al Glial Activation in Pilocarpine 2012documentin2010No ratings yet
- tmp37AD TMPDocument11 pagestmp37AD TMPFrontiersNo ratings yet
- Sarcoglycanopathies An Update 2021 Neuromuscular DisordersDocument7 pagesSarcoglycanopathies An Update 2021 Neuromuscular DisordersSuzie Simone Mardones SilvaNo ratings yet
- Sarcoglycanopathies An Update - 2021 - Neuromuscular DisordersDocument7 pagesSarcoglycanopathies An Update - 2021 - Neuromuscular DisordersSuzie Simone Mardones SilvaNo ratings yet
- Pancreatic β‑cell glutaminase 2 maintains glucose homeostasis under the condition of hyperglycaemiaDocument9 pagesPancreatic β‑cell glutaminase 2 maintains glucose homeostasis under the condition of hyperglycaemiascribdicinNo ratings yet
- Membrane Assembly and Morphogenesis: Drosophila Laminins Act As Key Regulators of BasementDocument12 pagesMembrane Assembly and Morphogenesis: Drosophila Laminins Act As Key Regulators of BasementPepeNo ratings yet
- Effects of Collagen IV On Neuroblastoma Cell Matrix-Related FunctionsDocument9 pagesEffects of Collagen IV On Neuroblastoma Cell Matrix-Related FunctionsdjackapNo ratings yet
- The Role of The Carbohydrates in Plasmatic MembranDocument26 pagesThe Role of The Carbohydrates in Plasmatic Membranmadwinyi skeptaNo ratings yet
- Kanno 2013Document11 pagesKanno 2013Ricardo EscNo ratings yet
- Serglycin Proteoglycan StorageDocument8 pagesSerglycin Proteoglycan StorageTeresa SilvaNo ratings yet
- Melatonin - Constant LightDocument11 pagesMelatonin - Constant LightTrần Khoa NguyênNo ratings yet
- LS VI Immuno Unit - 4.4Document28 pagesLS VI Immuno Unit - 4.4Masuri VarshaNo ratings yet
- Ivermectin GABA - Binding SiteDocument13 pagesIvermectin GABA - Binding SiteNoemiNo ratings yet
- Morphological and Physiological Interactions of NG2-glia With Astrocytes and NeuronsDocument10 pagesMorphological and Physiological Interactions of NG2-glia With Astrocytes and NeuronsRaffaello MobiliaNo ratings yet
- Dysferlin-Mediated Membrane Repair Protects The Heart From Stress-Induced Left Ventricular InjuryDocument9 pagesDysferlin-Mediated Membrane Repair Protects The Heart From Stress-Induced Left Ventricular InjuryBarbi CanalesNo ratings yet
- ArabidopsisDocument22 pagesArabidopsisRika Rahma PutriNo ratings yet
- Stabilizing Heterochromatin by DGCR8 Alleviates Senescence and OsteoarthritisDocument32 pagesStabilizing Heterochromatin by DGCR8 Alleviates Senescence and OsteoarthritisJacqueline Ramirez ArrietaNo ratings yet
- tmp2AF8 TMPDocument16 pagestmp2AF8 TMPFrontiersNo ratings yet
- SC 515409Document1 pageSC 515409juanfragarNo ratings yet
- Expanding The Phenotype of GMPPB Mutations: Doi:10.1093/brain/awv013 BRAIN 2015: 138 836-844 836Document9 pagesExpanding The Phenotype of GMPPB Mutations: Doi:10.1093/brain/awv013 BRAIN 2015: 138 836-844 836amraovcinaNo ratings yet
- AAV9 - s41591 019 0674 1Document39 pagesAAV9 - s41591 019 0674 1Felipe CourtNo ratings yet
- Rospect: Cellular BiochemistryDocument8 pagesRospect: Cellular BiochemistryRahmanu ReztaputraNo ratings yet
- cells: 17β-Estradiol Modulates SIRT1 and Halts Oxidative Stress-Mediated Cognitive Impairment in a Male Aging Mouse ModelDocument20 pagescells: 17β-Estradiol Modulates SIRT1 and Halts Oxidative Stress-Mediated Cognitive Impairment in a Male Aging Mouse ModelNAMIRA TANIYA TASYA TARADIFANo ratings yet
- GADD45B Growth Arrest and DNA-damage-inducible, BetaDocument6 pagesGADD45B Growth Arrest and DNA-damage-inducible, BetaHassan Ahmed HassanNo ratings yet
- Full-DikonversiDocument6 pagesFull-DikonversiJumi ApritasariNo ratings yet
- Bailey and Woodward, 1984 SC Cat Repression MutantsDocument6 pagesBailey and Woodward, 1984 SC Cat Repression MutantsAnaNo ratings yet
- Jove 122 55626Document9 pagesJove 122 55626Deliana Alexa Infante RojasNo ratings yet
- BAI1 Regulates Spatial Learning and Synaptic Plasticity in The HippocampusDocument12 pagesBAI1 Regulates Spatial Learning and Synaptic Plasticity in The HippocampusPeter TuNo ratings yet
- Dectin 1&2Document6 pagesDectin 1&2لؤي فارسNo ratings yet
- Formation of National Pride in The Educational Process. (In The Example of Abdullah Oripov's "Sahibqiron" Epic)Document4 pagesFormation of National Pride in The Educational Process. (In The Example of Abdullah Oripov's "Sahibqiron" Epic)Academic JournalNo ratings yet
- The Chemistry of Food: Proposed Sabbatical Program For Hwa Chong Institution (Year 2)Document2 pagesThe Chemistry of Food: Proposed Sabbatical Program For Hwa Chong Institution (Year 2)chuasioklengNo ratings yet
- Service Manual GSFS 5521 WS PDFDocument16 pagesService Manual GSFS 5521 WS PDFDEKOEBERNo ratings yet
- Data Mining: Sunitha R S Dept of ISE, RITDocument12 pagesData Mining: Sunitha R S Dept of ISE, RITSunitha Chetan R SNo ratings yet
- Bahasa InggrisDocument4 pagesBahasa Inggrisyaya keceNo ratings yet
- Career and Guidance ProgramDocument2 pagesCareer and Guidance ProgramCherry Mae Morales BandijaNo ratings yet
- Textiles in The World of SealingDocument2 pagesTextiles in The World of Sealingvadiraj2002No ratings yet
- 2 - 2 - 02 - 01 - 90 - 03 - Chlorination System Equipment Drawing - SchematicDocument24 pages2 - 2 - 02 - 01 - 90 - 03 - Chlorination System Equipment Drawing - SchematicABJ FarmNo ratings yet
- Orlander Text BookDocument783 pagesOrlander Text BookKai CaoNo ratings yet
- Curriculum Guide For Build ConstDocument72 pagesCurriculum Guide For Build ConstLuis VasquezNo ratings yet
- Application of HRM in 'The Social Network'Document9 pagesApplication of HRM in 'The Social Network'Atishay Jain100% (1)
- De-Mystifying Semiotics: Some Key Questions Answered: Rachel LawesDocument9 pagesDe-Mystifying Semiotics: Some Key Questions Answered: Rachel LawesminglessisNo ratings yet
- MTO Program For A BankDocument8 pagesMTO Program For A BankKamil FarooqNo ratings yet
- A'Bad, Karad, Nagpur Feasibility 22.11.2013Document3 pagesA'Bad, Karad, Nagpur Feasibility 22.11.2013Onal RautNo ratings yet
- 08.3. Theory of FlightDocument34 pages08.3. Theory of FlightRealyn LabaynaNo ratings yet
- Lecture 2 Engineering EconomicsDocument30 pagesLecture 2 Engineering Economicsahmer100% (1)
- Executive Summary of ApplichemDocument4 pagesExecutive Summary of ApplichemWen ZhangNo ratings yet
- Astronomy Education Around The WorldDocument4 pagesAstronomy Education Around The Worldhipoteticamente.contactoNo ratings yet
- Autocad Basic Course Outline:: Design By: Engr Hassan ShakoorDocument8 pagesAutocad Basic Course Outline:: Design By: Engr Hassan ShakoorLuqman AnsariNo ratings yet