You might also like
- Solutions To Exercises: - f3 f333 2V3f33U 3f3uu DDocument31 pagesSolutions To Exercises: - f3 f333 2V3f33U 3f3uu DLuis Miguel Angel Lizarraga MattoNo ratings yet
- Quantum Mechanics, Chapter 8Document7 pagesQuantum Mechanics, Chapter 8oneoonineNo ratings yet
- Simple Variation Method For The Hydrogen MoleculeDocument6 pagesSimple Variation Method For The Hydrogen Moleculetudor8sirbuNo ratings yet
- The Hydrogen Molecule Ion H2+Document5 pagesThe Hydrogen Molecule Ion H2+pappuNo ratings yet
- Solutions To Problem Set 2: P (E) + DPDocument12 pagesSolutions To Problem Set 2: P (E) + DPlantea1No ratings yet
- Lec26 AssDocument5 pagesLec26 AssFarid AkhtarNo ratings yet
- Aturan HuckelDocument12 pagesAturan HuckelMarvin ShadyNo ratings yet
- Foot Atomic Solutions by Zhao, C.Document32 pagesFoot Atomic Solutions by Zhao, C.Jacob Francis94% (16)
- Kronig Penney ModelDocument10 pagesKronig Penney ModelPhạm Hầu Thanh ViệtNo ratings yet
- QMChap 8Document9 pagesQMChap 8sanjeevchsNo ratings yet
- 2013 ps02 soUTIONlDocument12 pages2013 ps02 soUTIONlJerry ScottNo ratings yet
- Diq1m w8jrhDocument10 pagesDiq1m w8jrhMohsin MuhammadNo ratings yet
- Past Papers Solutions OutputDocument35 pagesPast Papers Solutions OutputCharlie Biopunk AlesNo ratings yet
- Solution20for20Midterm 201420fallDocument5 pagesSolution20for20Midterm 201420fall3.14159265No ratings yet
- Physics 12th Full Q&ADocument115 pagesPhysics 12th Full Q&AKiranshreeNo ratings yet
- Self-Consistent FieldDocument6 pagesSelf-Consistent FieldmekokiNo ratings yet
- ch8 ProbsDocument4 pagesch8 ProbsEkrem GüldesteNo ratings yet
- Introduction To Solid State Physics - Kittel, Charles - 8ed Solucionario PDFDocument61 pagesIntroduction To Solid State Physics - Kittel, Charles - 8ed Solucionario PDFmanuelomar87No ratings yet
- Wave Mechanics and The Schrödinger Equation (Chapter 7)Document8 pagesWave Mechanics and The Schrödinger Equation (Chapter 7)Abdul JabbarNo ratings yet
- P2214 Homework 14 Solutions - Spring 2011Document7 pagesP2214 Homework 14 Solutions - Spring 2011calcyeeNo ratings yet
- LCAO Band Structure Graphene CNTsDocument15 pagesLCAO Band Structure Graphene CNTsGbenga AgunbiadeNo ratings yet
- Excitons in Bulk and Low-Dimensional SemiconductorsDocument9 pagesExcitons in Bulk and Low-Dimensional Semiconductorsprakush_prakushNo ratings yet
- Solution Reif Cap6 7Document13 pagesSolution Reif Cap6 7Marcio Particheli100% (1)
- AppendixDocument21 pagesAppendixDayvid Mello NascimentoNo ratings yet
- MIT8 333F13 ExamReview3Document10 pagesMIT8 333F13 ExamReview3Henry De AriesNo ratings yet
- (Solutions) Kittel - Introduction To Solid State Physics 8Th EditionDocument60 pages(Solutions) Kittel - Introduction To Solid State Physics 8Th EditionCody Foster0% (1)
- UNIT-III-Band Theory of SolidsDocument8 pagesUNIT-III-Band Theory of SolidsGopichand surupulaNo ratings yet
- V V π 2 1 V π 2 V π 2Document3 pagesV V π 2 1 V π 2 V π 2jisteeleNo ratings yet
- 22.101 Applied Nuclear Physics (Fall 2006) Lecture 8 (10/4/06) Neutron-Proton ScatteringDocument7 pages22.101 Applied Nuclear Physics (Fall 2006) Lecture 8 (10/4/06) Neutron-Proton ScatteringmorganNo ratings yet
- Ij I I I I: 2 Jacob Lewis BourjailyDocument2 pagesIj I I I I: 2 Jacob Lewis BourjailySaul Perez BNo ratings yet
- Wave EquationDocument14 pagesWave EquationSawon KhanNo ratings yet
- Jest QuestionDocument16 pagesJest QuestionThirumugam SNo ratings yet
- 6 Hückel Theory: Basic AssumptionsDocument12 pages6 Hückel Theory: Basic AssumptionsAmran Mahfudh YatmeidhyNo ratings yet
- EM Waves in Material Media: 1 Wave PropagationDocument8 pagesEM Waves in Material Media: 1 Wave PropagationDinesh RamuNo ratings yet
- Nonrelativistic Limit of Dirac's Equation: 8.1 Large and Small ComponentsDocument4 pagesNonrelativistic Limit of Dirac's Equation: 8.1 Large and Small Componentspoecoek84No ratings yet
- Pchem10e Solutions Ch10 AtkinsonDocument32 pagesPchem10e Solutions Ch10 Atkinson동글No ratings yet
- Adrian Mulholland Molecular Structure and Bonding (II) SynopsisDocument12 pagesAdrian Mulholland Molecular Structure and Bonding (II) SynopsisAnni bannNo ratings yet
- Re Expansion On Compact Lie Groups 2019-02-15Document16 pagesRe Expansion On Compact Lie Groups 2019-02-15Rauan AkylzhanovNo ratings yet
- Townsend, Quantum Physics, CHAP - 4, 1DPotentialsDocument39 pagesTownsend, Quantum Physics, CHAP - 4, 1DPotentialsElcan DiogenesNo ratings yet
- Electric and Magnetic Fields Answers To Assignment 5 Q1:: 10 For FormulaDocument6 pagesElectric and Magnetic Fields Answers To Assignment 5 Q1:: 10 For FormulaShootingStarPhotonsNo ratings yet
- Thanks To Yossef and Shiang Yong For Their Input in This ProblemDocument8 pagesThanks To Yossef and Shiang Yong For Their Input in This ProblemIgnacio JuárezNo ratings yet
- CHAPTER 10: Atomic Structure and Atomic SpectraDocument25 pagesCHAPTER 10: Atomic Structure and Atomic SpectraVijay PradhanNo ratings yet
- Peskin QFT SolutionsDocument173 pagesPeskin QFT SolutionsAmir Iqbal100% (12)
- PHZ6426: Fall 2013 Problem Set # 6: Electron Transport. Phonons. Due Monday, 12/02 at The Time of The Class Instructor: D. L. Maslov Maslov@phys - Ufl.edu 392-0513 Rm. 2114 Office Hours: TR 3 pm-4 PMDocument9 pagesPHZ6426: Fall 2013 Problem Set # 6: Electron Transport. Phonons. Due Monday, 12/02 at The Time of The Class Instructor: D. L. Maslov Maslov@phys - Ufl.edu 392-0513 Rm. 2114 Office Hours: TR 3 pm-4 PMMkt SchrbrNo ratings yet
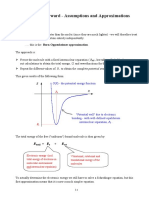
- 3 Assumptions and ApproximationsDocument5 pages3 Assumptions and ApproximationsJack RyderNo ratings yet
- Molecular Term SymbolsDocument5 pagesMolecular Term SymbolsAdam ButterworthNo ratings yet
- PHY491HW9SDocument3 pagesPHY491HW9SAsiri BandaraNo ratings yet
- Density of States in Graphene: Periodic Boundary ConditionDocument7 pagesDensity of States in Graphene: Periodic Boundary ConditionGemechis D DegagaNo ratings yet
- Kittel4 4Document3 pagesKittel4 4Gilberto PereiraNo ratings yet
- Solid State SatyabhamaDocument106 pagesSolid State Satyabhamawname384No ratings yet
- Tight Binding ProjectDocument12 pagesTight Binding ProjectNitish KumarNo ratings yet
- Pathria Solution1Document4 pagesPathria Solution1Mario Mede RiteNo ratings yet
- Hydrogen QMDocument20 pagesHydrogen QMAmanpreetNo ratings yet
- Atomic Physics ExamDocument3 pagesAtomic Physics Examvasudevan m.vNo ratings yet
- Tables of Coefficients for the Analysis of Triple Angular Correlations of Gamma-Rays from Aligned NucleiFrom EverandTables of Coefficients for the Analysis of Triple Angular Correlations of Gamma-Rays from Aligned NucleiNo ratings yet
- Ten-Decimal Tables of the Logarithms of Complex Numbers and for the Transformation from Cartesian to Polar Coordinates: Volume 33 in Mathematical Tables SeriesFrom EverandTen-Decimal Tables of the Logarithms of Complex Numbers and for the Transformation from Cartesian to Polar Coordinates: Volume 33 in Mathematical Tables SeriesNo ratings yet
- Advances in Structure Research by Diffraction Methods: Fortschritte der Strukturforschung mit BeugungsmethodenFrom EverandAdvances in Structure Research by Diffraction Methods: Fortschritte der Strukturforschung mit BeugungsmethodenR. BrillNo ratings yet
- Ridge Push and Slab PullDocument3 pagesRidge Push and Slab PullKatrina ChowNo ratings yet
- Thermal Properties of FoodsDocument12 pagesThermal Properties of FoodspedroloxxxNo ratings yet
- B787 Aft Fixed Galley. Swipe To See Close Range. Galley Inserts Have These Sizes ARINC Size 1, Arinc Size 2, Non-Arinc Size 3Document1 pageB787 Aft Fixed Galley. Swipe To See Close Range. Galley Inserts Have These Sizes ARINC Size 1, Arinc Size 2, Non-Arinc Size 3Husam FNo ratings yet
- Kailh CPG1511F01S172Document14 pagesKailh CPG1511F01S172xujiaqi730No ratings yet
- Air To Water Booster Unit Air To Water HEX UnitDocument13 pagesAir To Water Booster Unit Air To Water HEX Unittamago_fujiNo ratings yet
- Screening Machine RHEWUM WA 606d523ae0775Document2 pagesScreening Machine RHEWUM WA 606d523ae0775calvarez alvarezNo ratings yet
- Materials Today: Proceedings: Noor Suhailah Sulaiman, Muhammad Abbas Ahmad Zaini, Agus ArsadDocument4 pagesMaterials Today: Proceedings: Noor Suhailah Sulaiman, Muhammad Abbas Ahmad Zaini, Agus ArsadRaju BanikNo ratings yet
- Compressor Rotor Failure Due To FoulingDocument6 pagesCompressor Rotor Failure Due To FoulingAbdelkader TayebiNo ratings yet
- Refractometry and PolarimetryDocument2 pagesRefractometry and PolarimetryRichardNo ratings yet
- Cal Espes AutopipeDocument3 pagesCal Espes Autopipehernandez15No ratings yet
- Acha Et Al. 2015 PDFDocument73 pagesAcha Et Al. 2015 PDFJavier TolosanoNo ratings yet
- Heat Geek Mastery Workbook Home Print OffDocument101 pagesHeat Geek Mastery Workbook Home Print OffJason HaywardNo ratings yet
- Chapter 4 PDFDocument30 pagesChapter 4 PDFMuhammed Bn JihadNo ratings yet
- Topics: Text Book PHYSICS For Scientists and Engineers With Modern Physics (6 Ed) by Serway & JewettDocument84 pagesTopics: Text Book PHYSICS For Scientists and Engineers With Modern Physics (6 Ed) by Serway & JewettBala KumarNo ratings yet
- British Steel Universal Beams Ub Datasheet PDFDocument6 pagesBritish Steel Universal Beams Ub Datasheet PDFNicolae FloreaNo ratings yet
- Major Test: Hints & SolutionsDocument9 pagesMajor Test: Hints & SolutionsRahul AryanNo ratings yet
- Fit Up Report Format-1Document1 pageFit Up Report Format-1BalkishanDyavanapellyNo ratings yet
- Borehole ExtensometerDocument3 pagesBorehole ExtensometerAjay SinghNo ratings yet
- The Cast Oil Casting Case StudyDocument2 pagesThe Cast Oil Casting Case StudyAtta E Mustafa Mughal100% (1)
- Exercise 2 Thermodynamics Other Asic Concpts (Elements)Document13 pagesExercise 2 Thermodynamics Other Asic Concpts (Elements)Kamil GuillerganNo ratings yet
- RAYON Submittal - 12.8m - B1Document45 pagesRAYON Submittal - 12.8m - B1ahmed fathiNo ratings yet
- Hebat Sains Form 2 Changes of MatterDocument4 pagesHebat Sains Form 2 Changes of MatterHalizah RamthanNo ratings yet
- DSH 2umk031en03Document2 pagesDSH 2umk031en03agus sutiawanNo ratings yet
- Machine Elements 1: Assignment 2Document15 pagesMachine Elements 1: Assignment 2Jerald LatorreNo ratings yet
- Abedini (2020) - Optimum Design of Buckling-Restrained Braced FramesDocument14 pagesAbedini (2020) - Optimum Design of Buckling-Restrained Braced FramesGogyNo ratings yet
- 8187-116 Mechanical System DescriptionDocument5 pages8187-116 Mechanical System DescriptionJoelle SalamounNo ratings yet
- Air Conditioning and Refrigeration: Muhammad Bilal KhanDocument25 pagesAir Conditioning and Refrigeration: Muhammad Bilal KhanMuzammalRehmanNo ratings yet
- 2013 Micro Strcutral Transitional ZoneDocument8 pages2013 Micro Strcutral Transitional ZoneAvani DedhiaNo ratings yet
- Failure Analysis Case Study PDFDocument2 pagesFailure Analysis Case Study PDFScott50% (2)
- Magnaloy ProductsDocument223 pagesMagnaloy ProductsvankarpNo ratings yet