You might also like
- SPOT X DevDocument52 pagesSPOT X DevJaoNo ratings yet
- Kriging Method and ApplicationDocument56 pagesKriging Method and Applicationsisi100% (1)
- Ice Problem Sheet 1Document2 pagesIce Problem Sheet 1Muhammad Hamza AsgharNo ratings yet
- Alignment of SequencesDocument33 pagesAlignment of SequencesRaj Kumar SoniNo ratings yet
- Pairwise Sequence Alignment: CS 838 WWW - Cs.wisc - Edu/ Craven/cs838.html Mark Craven Craven@biostat - Wisc.edu January 2001Document18 pagesPairwise Sequence Alignment: CS 838 WWW - Cs.wisc - Edu/ Craven/cs838.html Mark Craven Craven@biostat - Wisc.edu January 2001Fadhili DungaNo ratings yet
- 02 AlignmentDocument52 pages02 AlignmentSiva AyyappanNo ratings yet
- FALLSEM2022-23 BIT3001 ETH VL2022230101828 Reference Material I 13-09-2022 MSADocument45 pagesFALLSEM2022-23 BIT3001 ETH VL2022230101828 Reference Material I 13-09-2022 MSATriparna PoddarNo ratings yet
- G P S Raghava: Basics of Sequence Alignment and Weight Matrices and DOT PlotDocument35 pagesG P S Raghava: Basics of Sequence Alignment and Weight Matrices and DOT PlotRaj Kumar SoniNo ratings yet
- Bioinformatics 1: Lecture 3 Key ConceptsDocument24 pagesBioinformatics 1: Lecture 3 Key ConceptsJean ReneNo ratings yet
- Sequence Alignment: Scoring MatricesDocument30 pagesSequence Alignment: Scoring MatricesSparsh NegiNo ratings yet
- Lecture2 3Document61 pagesLecture2 3Anirban SahaNo ratings yet
- Sequence Analysis - Pairwise AlignmentDocument26 pagesSequence Analysis - Pairwise AlignmentAnjana's WorldNo ratings yet
- PCB Lect02 Pairwise AllignDocument51 pagesPCB Lect02 Pairwise AllignLivsNo ratings yet
- Introduction To Bioinformatics: Sequence AlignmentDocument29 pagesIntroduction To Bioinformatics: Sequence AlignmentranjankumartiwariNo ratings yet
- Control Exp 8 Student ManualDocument7 pagesControl Exp 8 Student ManualSyed Nahid Ahmed TopuNo ratings yet
- Sequence Alignment 1Document24 pagesSequence Alignment 1Priyobrota ChyNo ratings yet
- frid seminarDocument30 pagesfrid seminarchahalamisha10No ratings yet
- Sequence Alignment Methods and AlgorithmsDocument37 pagesSequence Alignment Methods and Algorithmsapi-374725475% (4)
- Sequence Similarity: Motivation and MethodsDocument47 pagesSequence Similarity: Motivation and MethodsSomashree ChakrabortyNo ratings yet
- DNA AlignmentDocument76 pagesDNA AlignmentHammad HassanNo ratings yet
- Bioinformatics 1 - Lecture 8: Multiple Sequence AlignmentDocument20 pagesBioinformatics 1 - Lecture 8: Multiple Sequence AlignmentMohsan UllahNo ratings yet
- Sequence Alignment Lecture - Key ConceptsDocument19 pagesSequence Alignment Lecture - Key Conceptssk3 khanNo ratings yet
- Unit IiDocument14 pagesUnit IiDr. R. K. Selvakesavan PSGRKCWNo ratings yet
- Bioinformatics Algorithms and TechniquesDocument32 pagesBioinformatics Algorithms and TechniquesRohan RayNo ratings yet
- PDC Minor 2 - SlidesDocument190 pagesPDC Minor 2 - SlidesPiyush AmbulgekarNo ratings yet
- PDC Minor 2 - SlidesDocument190 pagesPDC Minor 2 - SlidesPiyush AmbulgekarNo ratings yet
- Lecture 5: Multiple Sequence Alignment: Introduction To Computational BiologyDocument34 pagesLecture 5: Multiple Sequence Alignment: Introduction To Computational BiologyJazmineNo ratings yet
- PAIR-WISE SEQUENCE COMPARISONDocument17 pagesPAIR-WISE SEQUENCE COMPARISONSubachandranNo ratings yet
- LAB No. 10 Analysis of Linear Control System Using Root Loci TechniqueDocument7 pagesLAB No. 10 Analysis of Linear Control System Using Root Loci TechniqueSanjar BeyNo ratings yet
- Analysis and Design of Cognitive Radio Networks and Distributed Radio Resource Management AlgorithmsDocument56 pagesAnalysis and Design of Cognitive Radio Networks and Distributed Radio Resource Management AlgorithmsVirendra TetarwalNo ratings yet
- Nyquist Plot and Stability Criteria - GATE Study Material in PDFDocument7 pagesNyquist Plot and Stability Criteria - GATE Study Material in PDFSHRIVENKATESHANo ratings yet
- Stat 401B Final Exam AnalysisDocument9 pagesStat 401B Final Exam AnalysisjuanEs2374pNo ratings yet
- Symmetry: Translations (Lattices)Document27 pagesSymmetry: Translations (Lattices)CJPATAGANNo ratings yet
- Tutorial Note 10 Midterm Solutions & Phylogenetic TreeDocument35 pagesTutorial Note 10 Midterm Solutions & Phylogenetic TreeRomario Tim VazNo ratings yet
- Root LocusDocument44 pagesRoot LocusDheer MehrotraNo ratings yet
- Design of Control Systems Cascade Root LocusDocument25 pagesDesign of Control Systems Cascade Root Locusaymanwolf3No ratings yet
- What is dynamic programming and how does it work? (39Document8 pagesWhat is dynamic programming and how does it work? (39Shadab Ahmed GhazalyNo ratings yet
- Root Locus Diagram - GATE Study Material in PDFDocument7 pagesRoot Locus Diagram - GATE Study Material in PDFAtul Choudhary100% (1)
- Principal Component AnalysisDocument7 pagesPrincipal Component Analysisdéborah_rosalesNo ratings yet
- RL en Espacios Con, Nuos: (Con Diaposi0vas de Ali Nouri)Document23 pagesRL en Espacios Con, Nuos: (Con Diaposi0vas de Ali Nouri)Kasey OwensNo ratings yet
- Root Locus Method 2Document33 pagesRoot Locus Method 2Umasankar ChilumuriNo ratings yet
- Encoding Information For DNA Computing: Shinnosuke SekiDocument45 pagesEncoding Information For DNA Computing: Shinnosuke SekiNNMSANo ratings yet
- Russell & Norvig Ch. 5: - Constraint Satisfaction Offers A Powerful Problem-Solving ParadigmDocument20 pagesRussell & Norvig Ch. 5: - Constraint Satisfaction Offers A Powerful Problem-Solving ParadigmirinarmNo ratings yet
- Daa 4.0 DP PDFDocument50 pagesDaa 4.0 DP PDFAbdus SadikNo ratings yet
- Root-Locus Design TechniquesDocument25 pagesRoot-Locus Design TechniquesNur DalilaNo ratings yet
- Root Locus Method 2Document33 pagesRoot Locus Method 2Patel DipenNo ratings yet
- 5.2 Root-Locus TechniqueDocument3 pages5.2 Root-Locus Techniquevikash yadavNo ratings yet
- Me 471 Phase Lead Root Locus ExampleDocument3 pagesMe 471 Phase Lead Root Locus ExampleCristian NapoleNo ratings yet
- Chapter 7 - Quantization Noise in DSPDocument31 pagesChapter 7 - Quantization Noise in DSPSarah KumakoNo ratings yet
- Factor Analysis with PASW: Exploring Common and Unique FactorsDocument40 pagesFactor Analysis with PASW: Exploring Common and Unique FactorsSaad MotawéaNo ratings yet
- Root Locus Diagram - GATE Study Material in PDFDocument7 pagesRoot Locus Diagram - GATE Study Material in PDFNarendra AgrawalNo ratings yet
- Point Pattern Analysis With Past: Øyvind Hammer, Natural History Museum, University of Oslo, 2011-01-03Document7 pagesPoint Pattern Analysis With Past: Øyvind Hammer, Natural History Museum, University of Oslo, 2011-01-03Emil TengwarNo ratings yet
- BLAST (Basic Local Alignment Search Tool)Document23 pagesBLAST (Basic Local Alignment Search Tool)Polu Chattopadhyay100% (1)
- Repetitive DNA Detection and Classification Using PILER and RepeatScoutDocument33 pagesRepetitive DNA Detection and Classification Using PILER and RepeatScoutPrabin KumarNo ratings yet
- ZRLocus For AnalysisDocument44 pagesZRLocus For AnalysisYusufANo ratings yet
- Feedback Control Systems Phase Plane AnalysisDocument9 pagesFeedback Control Systems Phase Plane AnalysisKthiha CnNo ratings yet
- Sequence Alignment 3Document43 pagesSequence Alignment 3Priyobrota ChyNo ratings yet
- Algorithms for Sequence AlignmentDocument37 pagesAlgorithms for Sequence AlignmentKrishnaNo ratings yet
- Z Transform PropertiesDocument12 pagesZ Transform Propertiesdebnathsuman49No ratings yet
- Measures of VariabilityDocument5 pagesMeasures of VariabilityMs. Mary UmitenNo ratings yet
- Simple Algebras, Base Change, and the Advanced Theory of the Trace Formula. (AM-120), Volume 120From EverandSimple Algebras, Base Change, and the Advanced Theory of the Trace Formula. (AM-120), Volume 120No ratings yet
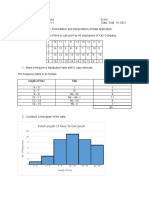
- Cedrix James Estoquia - OLLC Lesson 4.6 Presentation and Interpretation of Data ApplicationDocument4 pagesCedrix James Estoquia - OLLC Lesson 4.6 Presentation and Interpretation of Data ApplicationDeuK WR100% (1)
- Net CallDocument2 pagesNet CallFerdinand Monte Jr.100% (2)
- Impact of Government Expenditure On Human Capital Development (1990-2022)Document55 pagesImpact of Government Expenditure On Human Capital Development (1990-2022)malijessica907No ratings yet
- Basic Instructions: A Load (Contact) SymbolDocument3 pagesBasic Instructions: A Load (Contact) SymbolBaijayanti DasNo ratings yet
- ATC Flight PlanDocument21 pagesATC Flight PlanKabanosNo ratings yet
- Strategic ManagementDocument7 pagesStrategic ManagementSarah ShehataNo ratings yet
- Xerox University Microfilms: 300 North Zeeb Road Ann Arbor, Michigan 48106Document427 pagesXerox University Microfilms: 300 North Zeeb Road Ann Arbor, Michigan 48106Muhammad Haris Khan KhattakNo ratings yet
- Baghouse Compressed AirDocument17 pagesBaghouse Compressed Airmanh hung leNo ratings yet
- Jallikattu: Are Caste and Gender the Real Bulls to TameDocument67 pagesJallikattu: Are Caste and Gender the Real Bulls to TameMALLIKA NAGLENo ratings yet
- Valve Control System On A Venturi To Control FiO2 A Portable Ventilator With Fuzzy Logic Method Based On MicrocontrollerDocument10 pagesValve Control System On A Venturi To Control FiO2 A Portable Ventilator With Fuzzy Logic Method Based On MicrocontrollerIAES IJAINo ratings yet
- USP-NF 1251 Weighing On An Analytical BalanceDocument7 pagesUSP-NF 1251 Weighing On An Analytical BalanceZerish InaayaNo ratings yet
- DIK 18A Intan Suhariani 218112100 CJR Research MethodologyDocument9 pagesDIK 18A Intan Suhariani 218112100 CJR Research MethodologyFadli RafiNo ratings yet
- Eugene A. Nida - Theories of TranslationDocument15 pagesEugene A. Nida - Theories of TranslationYohanes Eko Portable100% (2)
- REHAU 20UFH InstallationDocument84 pagesREHAU 20UFH InstallationngrigoreNo ratings yet
- Schmitt Trigger FinalDocument4 pagesSchmitt Trigger Finalsidd14feb92No ratings yet
- IEC Developer version 6.n compiler error F1002Document6 pagesIEC Developer version 6.n compiler error F1002AnddyNo ratings yet
- Infineon IKCM30F60GD DataSheet v02 - 05 ENDocument17 pagesInfineon IKCM30F60GD DataSheet v02 - 05 ENBOOPATHIMANIKANDAN SNo ratings yet
- Indiana University PressDocument33 pagesIndiana University Pressrenato lopesNo ratings yet
- Professor Barry T Hart - BiographyDocument1 pageProfessor Barry T Hart - BiographyadelNo ratings yet
- SRFP 2015 Web List PDFDocument1 pageSRFP 2015 Web List PDFabhishekNo ratings yet
- ApplicationDocument11 pagesApplicationKamal Ra JNo ratings yet
- Ch1 - A Perspective On TestingDocument41 pagesCh1 - A Perspective On TestingcnshariffNo ratings yet
- A Guide For School LeadersDocument28 pagesA Guide For School LeadersIsam Al HassanNo ratings yet
- Limit of Outside Usage Outside Egypt ENDocument1 pageLimit of Outside Usage Outside Egypt ENIbrahem EmamNo ratings yet
- PROBLEM 7 - CDDocument4 pagesPROBLEM 7 - CDRavidya ShripatNo ratings yet
- Articulo SDocument11 pagesArticulo SGABRIELANo ratings yet
- (D-301) General Requirement in Equipment Installation - Rev.4 PDFDocument11 pages(D-301) General Requirement in Equipment Installation - Rev.4 PDFmihir_jha2No ratings yet
- Simha Lagna: First House Ruled by The Planet Sun (LEO) : The 1st House Known As The Ascendant orDocument3 pagesSimha Lagna: First House Ruled by The Planet Sun (LEO) : The 1st House Known As The Ascendant orRahulshah1984No ratings yet