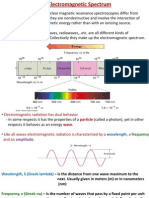
You might also like
- The Laws of Thermodynamics, A Very Short - Peter AtkinsDocument121 pagesThe Laws of Thermodynamics, A Very Short - Peter Atkinsae. xandp100% (1)
- IB HL Physics DefinitionsDocument7 pagesIB HL Physics DefinitionsshinyrayquazaNo ratings yet
- High Efficient Oxygen Reduction Performance of Fe Fe3C Nanopar - 2019 - Nano MatDocument6 pagesHigh Efficient Oxygen Reduction Performance of Fe Fe3C Nanopar - 2019 - Nano MatDuy-Khiem NguyenNo ratings yet
- Oriented Electron Transmission in Polyoxometalate-Metalloporphyrin Organic Framework For Highly Selective Electroreduction of CO2Document8 pagesOriented Electron Transmission in Polyoxometalate-Metalloporphyrin Organic Framework For Highly Selective Electroreduction of CO2Jam imtiazNo ratings yet
- Synthesis and Characterization of Nickel Ferrite Nanocatalysts For CO DecompositionDocument9 pagesSynthesis and Characterization of Nickel Ferrite Nanocatalysts For CO DecompositionJuancho PachonNo ratings yet
- Revisado 07Document9 pagesRevisado 07Juancho PachonNo ratings yet
- J Jcou 2019 04 010Document7 pagesJ Jcou 2019 04 010Roni GustiwaNo ratings yet
- 1 s2.0 S0360319921014336 MainDocument9 pages1 s2.0 S0360319921014336 Mainsatyajit beheraNo ratings yet
- Download Hierarchical Nico2O4 Cuo C Nanocomposite Derived From Copper Based Metal Organic Framework And Ni Co Hydroxides_ Excellent Electrocatalytic Activity Towards Methanol Oxidation Sara Sheikhi Fahimeh J full chapterDocument36 pagesDownload Hierarchical Nico2O4 Cuo C Nanocomposite Derived From Copper Based Metal Organic Framework And Ni Co Hydroxides_ Excellent Electrocatalytic Activity Towards Methanol Oxidation Sara Sheikhi Fahimeh J full chapterhenry.daughrity634100% (17)
- Elemental Mercury Capture From Flue Gas by Magnetic MN Fe Spinel: Effect of Chemical HeterogeneityDocument7 pagesElemental Mercury Capture From Flue Gas by Magnetic MN Fe Spinel: Effect of Chemical Heterogeneityyh hvNo ratings yet
- Asociacion Coreana de ElectroDocument8 pagesAsociacion Coreana de ElectroEspinosa RoNo ratings yet
- Light Olefins Synthesis From CO2 Hydrogenation Over Mixed FeeCoeK Supported On Micro Mesoporous Carbon CatalystsDocument15 pagesLight Olefins Synthesis From CO2 Hydrogenation Over Mixed FeeCoeK Supported On Micro Mesoporous Carbon CatalystsonglovelyableNo ratings yet
- Syngas Production Via Combined Steam and Carbon Dioxide Reforming of Methane Over Ni-Ce - MgAl2O4 Catalysts With Enhanced Coke ResistanceDocument7 pagesSyngas Production Via Combined Steam and Carbon Dioxide Reforming of Methane Over Ni-Ce - MgAl2O4 Catalysts With Enhanced Coke ResistanceWassachol SumarasinghaNo ratings yet
- Investigation of Highly Efficient Adsorbent Based On Ni-MOF-74 in TheDocument9 pagesInvestigation of Highly Efficient Adsorbent Based On Ni-MOF-74 in Thehamid saeedizadeNo ratings yet
- Dry Reforming of Methane Over The Cobalt Catalyst Theoretical Insights Into The Reaction Kinetics and Mechanism For Catalyst DeactivationDocument9 pagesDry Reforming of Methane Over The Cobalt Catalyst Theoretical Insights Into The Reaction Kinetics and Mechanism For Catalyst DeactivationJin WangNo ratings yet
- DFT Study of CO2 and H2O Co-Adsorption On CarbonDocument17 pagesDFT Study of CO2 and H2O Co-Adsorption On CarbonRafael Ricardo Celin ManceraNo ratings yet
- 1 s2.0 S0009261422005632 MainDocument7 pages1 s2.0 S0009261422005632 MainAamir ShafiqueNo ratings yet
- 10 1016@j Apsusc 2020 147047Document23 pages10 1016@j Apsusc 2020 147047Daniel MontalvoNo ratings yet
- Full Length Article: SciencedirectDocument12 pagesFull Length Article: SciencedirectAmir RahbariNo ratings yet
- 1 s2.0 S0926337323007622 MainDocument12 pages1 s2.0 S0926337323007622 Mainzhizheng wuNo ratings yet
- Fe MIL88BDocument9 pagesFe MIL88BThảo HàNo ratings yet
- Powder Technology: Nini Yuan, Hongcun Bai, Mei An, Jinpeng Zhang, Xiude Hu, Qingjie GuoDocument11 pagesPowder Technology: Nini Yuan, Hongcun Bai, Mei An, Jinpeng Zhang, Xiude Hu, Qingjie Guoyh hvNo ratings yet
- Daofeng 1-S2.0-S0255270117302271-MainDocument10 pagesDaofeng 1-S2.0-S0255270117302271-MainnadiaNo ratings yet
- 1 s2.0 S0360319921019510 MainDocument13 pages1 s2.0 S0360319921019510 MainSadegh AhmadiNo ratings yet
- Mondal JEC 15Document8 pagesMondal JEC 15Jay MandelNo ratings yet
- Materials Science & Engineering B: SciencedirectDocument6 pagesMaterials Science & Engineering B: SciencedirectRodrigoNo ratings yet
- CO2 Conversion To CO by Auto-Thermal Catalyst-Assisted Chemical LoopingDocument9 pagesCO2 Conversion To CO by Auto-Thermal Catalyst-Assisted Chemical LoopingNguyễn TuânNo ratings yet
- Pa Jpet Vol 01 Issue 01 - 09Document5 pagesPa Jpet Vol 01 Issue 01 - 09Isaac Hyuk DanielNo ratings yet
- Designing Perovskite Catalysts For Controlled Active-Site Exsolution in The Microwave Dry Reforming of MethaneDocument10 pagesDesigning Perovskite Catalysts For Controlled Active-Site Exsolution in The Microwave Dry Reforming of MethaneHoangNo ratings yet
- Eur. J. Inorg. Chem. 2017, 4982-4989Document8 pagesEur. J. Inorg. Chem. 2017, 4982-4989hungNo ratings yet
- 1 s2.0 S0169433221023795 MainDocument11 pages1 s2.0 S0169433221023795 MainDaniel MontalvoNo ratings yet
- Li 2017Document7 pagesLi 2017IscienceNo ratings yet
- Applied Catalysis B: Environmental: A. Ipek Paksoy, Burcu Selen Caglayan, A. Erhan AksoyluDocument11 pagesApplied Catalysis B: Environmental: A. Ipek Paksoy, Burcu Selen Caglayan, A. Erhan AksoyluleylaNo ratings yet
- Journal of Environmental Chemical EngineeringDocument10 pagesJournal of Environmental Chemical EngineeringDhanashree JagtapNo ratings yet
- Catalysis Today 2023 AbrhamDocument15 pagesCatalysis Today 2023 AbrhamboikoNo ratings yet
- Multimetallic MOFDocument8 pagesMultimetallic MOFLoga NathanNo ratings yet
- High-Performance Oxygen Evolution Catalyst Using Two-Dimensional Ultrathin Metal-Organic Frameworks NanosheetsDocument8 pagesHigh-Performance Oxygen Evolution Catalyst Using Two-Dimensional Ultrathin Metal-Organic Frameworks NanosheetsCB Dong SuwonNo ratings yet
- Novel Co-MOF/Graphene Oxide Electrocatalyst For Methanol OxidationDocument11 pagesNovel Co-MOF/Graphene Oxide Electrocatalyst For Methanol OxidationMuhammed AfnazNo ratings yet
- Accepted Manuscript: Journal of Photochemistry and Photobiology A: ChemistryDocument22 pagesAccepted Manuscript: Journal of Photochemistry and Photobiology A: Chemistry장위No ratings yet
- Preface For The Forum On Metal Organic Frameworks For Energy ApplicationsDocument3 pagesPreface For The Forum On Metal Organic Frameworks For Energy ApplicationsTHI PHAM TANNo ratings yet
- Materials Letters: Moxin Yu, Li Zhang, Xiaojun He, Huanhuan Yu, Jiufeng Han, Mingbo WuDocument4 pagesMaterials Letters: Moxin Yu, Li Zhang, Xiaojun He, Huanhuan Yu, Jiufeng Han, Mingbo WuBreenteeNo ratings yet
- Método de Coprecipitación MnFeMoO4Document9 pagesMétodo de Coprecipitación MnFeMoO4Alifhers Salim Mestra AcostaNo ratings yet
- Full Length Article: SciencedirectDocument9 pagesFull Length Article: Sciencedirectyh hvNo ratings yet
- Journal of Electroanalytical Chemistry: SciencedirectDocument7 pagesJournal of Electroanalytical Chemistry: Sciencedirectvijayamathubalan pandyNo ratings yet
- Density Functional Theory Study of Adsorption and Dissociation of CH2Cl2 OnDocument14 pagesDensity Functional Theory Study of Adsorption and Dissociation of CH2Cl2 OnLarissa VourlisNo ratings yet
- Atomically Dispersed Manganese Catalsyt For ORRDocument11 pagesAtomically Dispersed Manganese Catalsyt For ORRShibil AhamedNo ratings yet
- Carbonic Anhydrase Immobilized On Encapsulated Magnetic Nanoparticles For CO2 SequestrationDocument8 pagesCarbonic Anhydrase Immobilized On Encapsulated Magnetic Nanoparticles For CO2 SequestrationASinopoliNo ratings yet
- Enhancement Low Temperature NH3 SCR Activity of The Fe MN Mo - 2023 - MolecularDocument11 pagesEnhancement Low Temperature NH3 SCR Activity of The Fe MN Mo - 2023 - MolecularDana MateiNo ratings yet
- Baranwal-PrasannaVenkatesh2017 Article InvestigationOfCarbonSteelAnod PDFDocument12 pagesBaranwal-PrasannaVenkatesh2017 Article InvestigationOfCarbonSteelAnod PDFestiven ValderramaNo ratings yet
- Computational and Theoretical Chemistry: SciencedirectDocument8 pagesComputational and Theoretical Chemistry: SciencedirectAna LuizaNo ratings yet
- Zeolitic-Imidazolate-Framework-Derived Co@Co3O4 Embedded IntoDocument9 pagesZeolitic-Imidazolate-Framework-Derived Co@Co3O4 Embedded IntoXINFENG ZHUNo ratings yet
- 基于胶体晶体模板化方法的化学循环制氢的改进氧化铁氧载体Document10 pages基于胶体晶体模板化方法的化学循环制氢的改进氧化铁氧载体yh hvNo ratings yet
- Perovskite Alcohol ReformingDocument14 pagesPerovskite Alcohol ReformingposidontubeNo ratings yet
- Improved Methanol Yield and Selectivity From CO2 Hydrogenation Using A Novel Cu-ZnO-ZrO2 Catalyst Supported On Mg-Al Layered Double Hydroxide (LDH)Document8 pagesImproved Methanol Yield and Selectivity From CO2 Hydrogenation Using A Novel Cu-ZnO-ZrO2 Catalyst Supported On Mg-Al Layered Double Hydroxide (LDH)Nguyễn TuânNo ratings yet
- La0 6sr0 4co0 2fe0 8o3 Perovskite A Stable Anode Catalyst For Direct Methane Solid Oxide Fuel CellsDocument17 pagesLa0 6sr0 4co0 2fe0 8o3 Perovskite A Stable Anode Catalyst For Direct Methane Solid Oxide Fuel CellsG XeragoNo ratings yet
- Hamou D 2019Document10 pagesHamou D 2019Adriano Aj-formataçãoNo ratings yet
- Energies 17 01483Document12 pagesEnergies 17 01483Maher AzzouzNo ratings yet
- Improvement in VRFB by Modified Graphite Felt - Mahanta - 2020Document13 pagesImprovement in VRFB by Modified Graphite Felt - Mahanta - 2020SureshBharadwajNo ratings yet
- Pagination MTCHEM 101716Document10 pagesPagination MTCHEM 101716없다이름No ratings yet
- Guo 2010Document6 pagesGuo 2010yh hvNo ratings yet
- Chemical Engineering Journal: A B C D e e CDocument10 pagesChemical Engineering Journal: A B C D e e CHemanth Peddavenkatappa GariNo ratings yet
- 汞的净化0 湿法氧化烟气及其机理研究进展Document14 pages汞的净化0 湿法氧化烟气及其机理研究进展yh hvNo ratings yet
- Fuel Processing Technology: Jinchen Ma, Xin Tian, Bo Zhao, Xiaoshan Li, Yongchun Zhao, Haibo Zhao, Chuguang ZhengDocument11 pagesFuel Processing Technology: Jinchen Ma, Xin Tian, Bo Zhao, Xiaoshan Li, Yongchun Zhao, Haibo Zhao, Chuguang Zhengyh hvNo ratings yet
- Mini Review: Controlling Lattice Oxygen Activity of Oxygen Carrier Materials by Design: A Review and PerspectiveDocument11 pagesMini Review: Controlling Lattice Oxygen Activity of Oxygen Carrier Materials by Design: A Review and Perspectiveyh hvNo ratings yet
- 预热 燃烧耦合工艺预热阶段煤氮转化的实验与动力学研究Document8 pages预热 燃烧耦合工艺预热阶段煤氮转化的实验与动力学研究yh hvNo ratings yet
- Fuel Processing Technology: Yingju Yang, Jing Liu, Zhen Wang, Feng LiuDocument6 pagesFuel Processing Technology: Yingju Yang, Jing Liu, Zhen Wang, Feng Liuyh hvNo ratings yet
- Structural, Chemical, and Magnetic Investigations of Core Shell Zinc Ferrite NanoparticlesDocument11 pagesStructural, Chemical, and Magnetic Investigations of Core Shell Zinc Ferrite Nanoparticlesyh hvNo ratings yet
- Powder Technology: Nini Yuan, Hongcun Bai, Mei An, Jinpeng Zhang, Xiude Hu, Qingjie GuoDocument11 pagesPowder Technology: Nini Yuan, Hongcun Bai, Mei An, Jinpeng Zhang, Xiude Hu, Qingjie Guoyh hvNo ratings yet
- 核壳结构镍基氧载体上固体燃料化学循环燃烧的还原和氧化动力学:已开发的粒度分布模型的应用Document8 pages核壳结构镍基氧载体上固体燃料化学循环燃烧的还原和氧化动力学:已开发的粒度分布模型的应用yh hvNo ratings yet
- Effect of Oxygen Vacancy Defect On The Magnetic Properties of Co-Doped ZnoDocument7 pagesEffect of Oxygen Vacancy Defect On The Magnetic Properties of Co-Doped Znoyh hvNo ratings yet
- Chemical Engineering Journal: Yingju Yang, Jing Liu, Bingkai Zhang, Feng LiuDocument7 pagesChemical Engineering Journal: Yingju Yang, Jing Liu, Bingkai Zhang, Feng Liuyh hvNo ratings yet
- Effects of No, A-Fe O, G-Fe O, and HCL On Mercury Transformations in A 7-Kw Coal Combustion SystemDocument20 pagesEffects of No, A-Fe O, G-Fe O, and HCL On Mercury Transformations in A 7-Kw Coal Combustion Systemyh hvNo ratings yet
- Elemental Mercury Capture From Flue Gas by Magnetic MN Fe Spinel: Effect of Chemical HeterogeneityDocument7 pagesElemental Mercury Capture From Flue Gas by Magnetic MN Fe Spinel: Effect of Chemical Heterogeneityyh hvNo ratings yet
- Guo 2010Document6 pagesGuo 2010yh hvNo ratings yet
- Mercury Removal From Flue Gases by Novel Regenerable Magnetic Nanocomposite SorbentsDocument6 pagesMercury Removal From Flue Gases by Novel Regenerable Magnetic Nanocomposite Sorbentsyh hvNo ratings yet
- 基于胶体晶体模板化方法的化学循环制氢的改进氧化铁氧载体Document10 pages基于胶体晶体模板化方法的化学循环制氢的改进氧化铁氧载体yh hvNo ratings yet
- Materials Characterization: SciencedirectDocument11 pagesMaterials Characterization: Sciencedirectyh hvNo ratings yet
- 10.1016/j.cis.2016.04.001: Advances in Colloid and Interface ScienceDocument69 pages10.1016/j.cis.2016.04.001: Advances in Colloid and Interface Scienceyh hvNo ratings yet
- 从煤合成气中去除元素泰汞 使用磁性茶 生物炭Document12 pages从煤合成气中去除元素泰汞 使用磁性茶 生物炭yh hvNo ratings yet
- Full Length Article: SciencedirectDocument9 pagesFull Length Article: Sciencedirectyh hvNo ratings yet
- MSC ChemistryDocument52 pagesMSC Chemistryanon_30148465No ratings yet
- Chap 12 - IRDocument30 pagesChap 12 - IRTanvir AhmedNo ratings yet
- Rectifier Applications: HandbookDocument272 pagesRectifier Applications: Handbookjhvfbuio5485No ratings yet
- Uv VisablespectroscopyatheerDocument18 pagesUv Visablespectroscopyatheerareej juttNo ratings yet
- Workbook Answers: Exercise 2.1Document3 pagesWorkbook Answers: Exercise 2.1Akhmad Nur100% (1)
- Chapter 6Document20 pagesChapter 6alyalaswad4445No ratings yet
- 1 Atomic Structure PDFDocument20 pages1 Atomic Structure PDFanilkumarsharma1969No ratings yet
- Course Code: PHA 305 UV-Visible SpectrophotometryDocument38 pagesCourse Code: PHA 305 UV-Visible SpectrophotometryMahadi Hasan KhanNo ratings yet
- Quantum Numbers: N, L, M, and SDocument9 pagesQuantum Numbers: N, L, M, and Seminent career academyNo ratings yet
- Excited States and Photochemistry of Organic Molecules Klessinger M Michl J VCH 1995 PDFDocument281 pagesExcited States and Photochemistry of Organic Molecules Klessinger M Michl J VCH 1995 PDFpersiandrunkardNo ratings yet
- Quantum Mechanics TutorialDocument6 pagesQuantum Mechanics TutorialAnonymous ziJjOnGnNo ratings yet
- CaliceneDocument9 pagesCaliceneAndrew BirdNo ratings yet
- ChemistryDocument23 pagesChemistryM KamranNo ratings yet
- Science 9 - Q2 - Week1 - Melc 1-3Document18 pagesScience 9 - Q2 - Week1 - Melc 1-3Rhyan Zero-four BaluyutNo ratings yet
- 7-Dispersing LNP US20120228565A1Document15 pages7-Dispersing LNP US20120228565A1Jamie RobinsonNo ratings yet
- Chemistry Notes 1 - Atomic Model and Atomic Theory Frame NotesDocument7 pagesChemistry Notes 1 - Atomic Model and Atomic Theory Frame NotesErin KabezNo ratings yet
- Caltech RefDocument308 pagesCaltech RefSukrit ChatterjeeNo ratings yet
- ELEC3908F16W17Notes PDFDocument149 pagesELEC3908F16W17Notes PDFXman099450% (2)
- SJPO Special Round 2009 Sample PDFDocument8 pagesSJPO Special Round 2009 Sample PDFziwei_from_chinaNo ratings yet
- Science 9 Quarter 2 Week 2 Bohrs Model vs. Quantum Mechanical ModelDocument40 pagesScience 9 Quarter 2 Week 2 Bohrs Model vs. Quantum Mechanical ModelMimoNo ratings yet
- Lakhmir Singh Solutions For Class 9 Feb22 Chemistry Chapter 4 Structure of AtomDocument14 pagesLakhmir Singh Solutions For Class 9 Feb22 Chemistry Chapter 4 Structure of AtomDarshilNo ratings yet
- Practice QuestionsDocument26 pagesPractice QuestionsL Michelle MackNo ratings yet
- Physical Sciences Lesson 2 The Atomic Structure and The Chemical ElementsDocument11 pagesPhysical Sciences Lesson 2 The Atomic Structure and The Chemical ElementsJustin Bird100% (1)
- FTIR Lecture SlidesDocument25 pagesFTIR Lecture Slidesabdul rehman khanNo ratings yet
- Term Paper 13251Document9 pagesTerm Paper 13251Raghav ChaudharyNo ratings yet
- 2.3 Developments Leading To The Bohr'S Model of AtomDocument29 pages2.3 Developments Leading To The Bohr'S Model of AtomAbijithGamer123No ratings yet
- Q2 WLP Sci9 Week1Document7 pagesQ2 WLP Sci9 Week1Ronelyn SobrianoNo ratings yet
- Zeeman EffectDocument10 pagesZeeman EffectSayyed SalmanNo ratings yet