You might also like
- Coal Science: Volume 1From EverandCoal Science: Volume 1Martin L. GorbatyNo ratings yet
- 1 s2.0 S0926337323007622 MainDocument12 pages1 s2.0 S0926337323007622 Mainzhizheng wuNo ratings yet
- Recent Progress in Syngas Production Via Catalytic CO2 Hydrogenation ReactionDocument11 pagesRecent Progress in Syngas Production Via Catalytic CO2 Hydrogenation Reactionkishore dasmunshiNo ratings yet
- 5253 FTPDocument3 pages5253 FTPAviad CahanaNo ratings yet
- Journal of Catalysis: Ang Cao, Zhenbin Wang, Hao Li, Ahmed O. Elnabawy, Jens K. NørskovDocument7 pagesJournal of Catalysis: Ang Cao, Zhenbin Wang, Hao Li, Ahmed O. Elnabawy, Jens K. NørskovDavid KehindeNo ratings yet
- A DFT Investigation of The Mechanisms of CO2 and CO Methanation On FeDocument7 pagesA DFT Investigation of The Mechanisms of CO2 and CO Methanation On FeHoa Hướng Dương HnueNo ratings yet
- 9969 33653 2 PBDocument14 pages9969 33653 2 PBTirtana PrasetiaNo ratings yet
- J Jcou 2019 04 010Document7 pagesJ Jcou 2019 04 010Roni GustiwaNo ratings yet
- Liu 2003Document13 pagesLiu 2003Eko NevrianNo ratings yet
- Reichenbach 2018Document7 pagesReichenbach 2018Neha NegiNo ratings yet
- Co2 Activation Over Catalytic Surfaces: Chemphyschem August 2017Document10 pagesCo2 Activation Over Catalytic Surfaces: Chemphyschem August 2017sheraz ahmadNo ratings yet
- Zeolite Fixed Cobalt - Nickel Nanoparticles For Coking and Sintering Resistance in Dry Reforming of MethaneDocument10 pagesZeolite Fixed Cobalt - Nickel Nanoparticles For Coking and Sintering Resistance in Dry Reforming of MethaneJulieta StassiNo ratings yet
- Activated CarbonDocument7 pagesActivated CarbonmghasdiNo ratings yet
- Improved Methanol Yield and Selectivity From CO2 Hydrogenation Using A Novel Cu-ZnO-ZrO2 Catalyst Supported On Mg-Al Layered Double Hydroxide (LDH)Document8 pagesImproved Methanol Yield and Selectivity From CO2 Hydrogenation Using A Novel Cu-ZnO-ZrO2 Catalyst Supported On Mg-Al Layered Double Hydroxide (LDH)Nguyễn TuânNo ratings yet
- Electrochemical Reduction of CO Catalyzed by Metal NanocatalystsDocument12 pagesElectrochemical Reduction of CO Catalyzed by Metal NanocatalystsaliNo ratings yet
- 1 s2.0 S0169433221023795 MainDocument11 pages1 s2.0 S0169433221023795 MainDaniel MontalvoNo ratings yet
- DFT Study of CO2 and H2O Co-Adsorption On CarbonDocument17 pagesDFT Study of CO2 and H2O Co-Adsorption On CarbonRafael Ricardo Celin ManceraNo ratings yet
- Density Functional Theory Study of Adsorption and Dissociation of CH2Cl2 OnDocument14 pagesDensity Functional Theory Study of Adsorption and Dissociation of CH2Cl2 OnLarissa VourlisNo ratings yet
- Cat Al 10080944Document25 pagesCat Al 10080944Nina NinaNo ratings yet
- On The General Mechanism of Photocatalytic Reduction of CO2Document10 pagesOn The General Mechanism of Photocatalytic Reduction of CO2alejandro sifuentes clementeNo ratings yet
- 10 1016@j Jphotochem 2019 111933Document10 pages10 1016@j Jphotochem 2019 111933zmaxprom5No ratings yet
- Art. 2 AdelantoDocument14 pagesArt. 2 AdelantoYuliiana HernandezNo ratings yet
- Low Temperature Catalytic Reverse Water-Gas Shift ReactionDocument14 pagesLow Temperature Catalytic Reverse Water-Gas Shift ReactionRen FengNo ratings yet
- Lim2009 PDFDocument8 pagesLim2009 PDFNguyen Van DuyNo ratings yet
- A Short Review of CatalysisDocument12 pagesA Short Review of CatalysisEliezer BecziNo ratings yet
- Bore Ribo On 2018Document8 pagesBore Ribo On 2018Hêny CarlênicNo ratings yet
- Electrical and Co Gas Sensing Properties of Nanostructured La Ce Coo Perovskite Prepared by Activated Reactive SynthesisDocument9 pagesElectrical and Co Gas Sensing Properties of Nanostructured La Ce Coo Perovskite Prepared by Activated Reactive SynthesisAmir GhasdiNo ratings yet
- Wu 2020Document7 pagesWu 2020Daniel MontalvoNo ratings yet
- CC SynthesisDocument9 pagesCC SynthesisseenuRaviNo ratings yet
- 1 s2.0 S0304389420320847 MainDocument9 pages1 s2.0 S0304389420320847 MainclaudiacarranzafNo ratings yet
- Formic Acid SynthesisDocument7 pagesFormic Acid SynthesisJoeNo ratings yet
- Journal of Environmental Chemical Engineering: 2 2 Yan Resing Dias, Oscar W. Perez-LopezDocument8 pagesJournal of Environmental Chemical Engineering: 2 2 Yan Resing Dias, Oscar W. Perez-Lopezfarah al-sudaniNo ratings yet
- 1 s2.0 S0009261422005632 MainDocument7 pages1 s2.0 S0009261422005632 MainAamir ShafiqueNo ratings yet
- Engineering Pt-Mn2O3 Interface To Boost Selective Oxidation of Ethylene Glycol To Glycolic AcidDocument11 pagesEngineering Pt-Mn2O3 Interface To Boost Selective Oxidation of Ethylene Glycol To Glycolic Acid63011373No ratings yet
- Performance Prediction and Evaluation of CO2 Utilization With Conjoined Electrolysis and Carbonation Using Desalinated Rejected Seawater BrineDocument9 pagesPerformance Prediction and Evaluation of CO2 Utilization With Conjoined Electrolysis and Carbonation Using Desalinated Rejected Seawater BrineJHON JAIRO RAMOS FLOREZNo ratings yet
- Journal of Natural Gas Science and Engineering: Yuefa Wang, Zhongxi Chao, Hugo A. JakobsenDocument9 pagesJournal of Natural Gas Science and Engineering: Yuefa Wang, Zhongxi Chao, Hugo A. JakobsenDaniel Z JucaNo ratings yet
- Catalysts 12 00403 v2Document33 pagesCatalysts 12 00403 v2Châu Tinh TổNo ratings yet
- High CO2 Absorption Capacity of Metal Based Ionic LiquidsDocument8 pagesHigh CO2 Absorption Capacity of Metal Based Ionic Liquidspathakshivam767No ratings yet
- Dutta 2012Document5 pagesDutta 2012Marcelo DutraNo ratings yet
- High Temperature Co2 ReactionsDocument17 pagesHigh Temperature Co2 Reactions21bmc015No ratings yet
- Chemical Engineering ScienceDocument14 pagesChemical Engineering ScienceAzharuddin_kfupmNo ratings yet
- Designing Perovskite Catalysts For Controlled Active-Site Exsolution in The Microwave Dry Reforming of MethaneDocument10 pagesDesigning Perovskite Catalysts For Controlled Active-Site Exsolution in The Microwave Dry Reforming of MethaneHoangNo ratings yet
- 10 1016@j Jcat 2020 01 028Document7 pages10 1016@j Jcat 2020 01 028Gonzalo BenavidesNo ratings yet
- CO Oxidation Over PD Supported Catalysts - in Situ Study of The ElectricDocument8 pagesCO Oxidation Over PD Supported Catalysts - in Situ Study of The ElectricPatrick Tejada AdriazolaNo ratings yet
- 1 AntoniusDocument6 pages1 Antoniusapi-3728640No ratings yet
- 2020 PDFDocument8 pages2020 PDFlala habibiNo ratings yet
- RH Catalyzed FormylationDocument4 pagesRH Catalyzed FormylationHimadri SahaNo ratings yet
- Donazzi 2008Document18 pagesDonazzi 2008Monika MartinezNo ratings yet
- Carbonation-Calcination Cycle Using High Reactivity Calcium Oxide For Carbon Dioxide Separation From Flue GasDocument8 pagesCarbonation-Calcination Cycle Using High Reactivity Calcium Oxide For Carbon Dioxide Separation From Flue GasPola PoliNo ratings yet
- Koh ActivationDocument9 pagesKoh ActivationAloneNo ratings yet
- Catalysts: Catalytic Hydrogen Production From Methane: A Review On Recent Progress and ProspectDocument18 pagesCatalysts: Catalytic Hydrogen Production From Methane: A Review On Recent Progress and ProspectMuhammad HarisNo ratings yet
- Solid State Ionics: Tae Ho Shin, Jae-Ha Myung, Khan M. Naeem, Cristian Savaniu, John T.S. IrvineDocument4 pagesSolid State Ionics: Tae Ho Shin, Jae-Ha Myung, Khan M. Naeem, Cristian Savaniu, John T.S. Irvinedennisitty01No ratings yet
- 10 1016@j Apsusc 2020 147047Document23 pages10 1016@j Apsusc 2020 147047Daniel MontalvoNo ratings yet
- CO2 Splitting To Carbon - Ceria - Nat CommunDocument8 pagesCO2 Splitting To Carbon - Ceria - Nat CommunherayatiNo ratings yet
- Applied Clay Science: Research PaperDocument9 pagesApplied Clay Science: Research Papermaryam Saket OsgoueiNo ratings yet
- 2009 - Catalytic Conversion of NaHCO3 Into Formic Acid in Mild Hydrothermal Conditions For Co2 UtilizationDocument6 pages2009 - Catalytic Conversion of NaHCO3 Into Formic Acid in Mild Hydrothermal Conditions For Co2 UtilizationMaría Andérez FernándezNo ratings yet
- Modeling The Adsorption of CO On Small PT, Fe and Co Clusters For The Fischer-Tropsch SynthesisDocument14 pagesModeling The Adsorption of CO On Small PT, Fe and Co Clusters For The Fischer-Tropsch SynthesisLilian PaesNo ratings yet
- Highly Stable M/Nioemgo (M Co, Cu and Fe) Catalysts Towards Co MethanationDocument16 pagesHighly Stable M/Nioemgo (M Co, Cu and Fe) Catalysts Towards Co Methanationfarah al-sudaniNo ratings yet
- 1 s2.0 S2468111320300426 MainDocument11 pages1 s2.0 S2468111320300426 Main21bmc015No ratings yet
- Nanomaterials 11 03265 v3Document40 pagesNanomaterials 11 03265 v3Wassachol SumarasinghaNo ratings yet
- Wang Et Al-2004-Polymer InternationalDocument10 pagesWang Et Al-2004-Polymer InternationalJin WangNo ratings yet
- NanodispersedDocument9 pagesNanodispersedJin WangNo ratings yet
- Debecker Et Al-2009-Chemistry - A European JournalDocument16 pagesDebecker Et Al-2009-Chemistry - A European JournalJin WangNo ratings yet
- Ma Et Al. - Unknown - Exfoliating Layered Double Hydroxides in Formamide A Method To Obtain Positively Charged Nanosheets-AnnotatedDocument5 pagesMa Et Al. - Unknown - Exfoliating Layered Double Hydroxides in Formamide A Method To Obtain Positively Charged Nanosheets-AnnotatedJin WangNo ratings yet
- Cordeiro Et Al. - 2012 - LDHs Instability in Esterification Reactions and Their Conversion To Catalytically Active Layered Carboxylates-AnnotatedDocument8 pagesCordeiro Et Al. - 2012 - LDHs Instability in Esterification Reactions and Their Conversion To Catalytically Active Layered Carboxylates-AnnotatedJin WangNo ratings yet
- A New Zinc Hydroxide Nitrate HeterogeneoDocument5 pagesA New Zinc Hydroxide Nitrate HeterogeneoJin WangNo ratings yet
- NETAFIM Drip Irrigation System HandbookDocument96 pagesNETAFIM Drip Irrigation System HandbookCarlos Rovello GandoNo ratings yet
- IPS d-SIGN 98Document2 pagesIPS d-SIGN 98Toma IoanaNo ratings yet
- Common Errors in Hydronic Radiant Panel Heating Systems John-SiegenthalerDocument35 pagesCommon Errors in Hydronic Radiant Panel Heating Systems John-SiegenthalerJM100% (1)
- Enve 208 Experiment 2Document9 pagesEnve 208 Experiment 2mihrican3020% (1)
- ActionTypes LookupDocument2 pagesActionTypes LookupRohitKumarNo ratings yet
- Gate Valves: Cast Iron Gate Valve Class 125Document1 pageGate Valves: Cast Iron Gate Valve Class 125Daniel SanNo ratings yet
- Casing: Conductor Surface Intermediate Liner ProductionDocument8 pagesCasing: Conductor Surface Intermediate Liner ProductionMostafa IbrahimNo ratings yet
- MWM TCG2020Document8 pagesMWM TCG2020Md Iqbal HossainNo ratings yet
- In Vitro Dissolution Testing Models: Seminar OnDocument23 pagesIn Vitro Dissolution Testing Models: Seminar OnPrashant GaikwadNo ratings yet
- CV - Chemical EngineeringDocument2 pagesCV - Chemical EngineeringsohanNo ratings yet
- Piping Spool Fabrication ProcedureDocument14 pagesPiping Spool Fabrication ProcedureMiky Andrean100% (6)
- Grundfosliterature 5235678Document104 pagesGrundfosliterature 5235678Adriana SandruNo ratings yet
- DD Cen Ts 1992-4-2009 Design of Fastenings For Use in ConcreteDocument190 pagesDD Cen Ts 1992-4-2009 Design of Fastenings For Use in ConcretemirosekNo ratings yet
- Pratham International School: Subject: Science Grade: VDocument2 pagesPratham International School: Subject: Science Grade: VBharath BabuNo ratings yet
- Flexonyl Blue WF 153Document2 pagesFlexonyl Blue WF 153Leandro EsvizaNo ratings yet
- Assignment On: Cultivation Practices of BajraDocument26 pagesAssignment On: Cultivation Practices of BajraAbhishek kumarNo ratings yet
- F-Block ElementsDocument8 pagesF-Block ElementsSai Sasivardhan GampaNo ratings yet
- Sample WPS OQRDocument4 pagesSample WPS OQRavgpaulNo ratings yet
- A Gyermekek Gyógynövényes KezeléseDocument333 pagesA Gyermekek Gyógynövényes KezeléseShakti Ayurveda VácNo ratings yet
- Foe Sample QuistionSDocument21 pagesFoe Sample QuistionSYuGenNo ratings yet
- Image Receptors: Dr. Chona D. CabatayDocument37 pagesImage Receptors: Dr. Chona D. CabatayEzekiel MartinezNo ratings yet
- Mildew (Fungus) Resistance of Paper and Paperboard: Standard Test Methods ForDocument4 pagesMildew (Fungus) Resistance of Paper and Paperboard: Standard Test Methods ForShaker QaidiNo ratings yet
- Metronidazole AssyDocument2 pagesMetronidazole AssysaiNo ratings yet
- Eudiometry:: Ans. SolDocument2 pagesEudiometry:: Ans. Soltan jigNo ratings yet
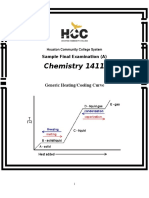
- Chemistry 1411: Generic Heating/Cooling CurveDocument19 pagesChemistry 1411: Generic Heating/Cooling CurveKinal PatelNo ratings yet
- Convac 3000P/PQ Vacuum Cleaner: InstructionsDocument3 pagesConvac 3000P/PQ Vacuum Cleaner: InstructionsIacob VioletaNo ratings yet
- Pressurized Dosage FormsDocument85 pagesPressurized Dosage FormsHuma Hameed Dogar100% (1)
- Periodic Table of The ElementsDocument4 pagesPeriodic Table of The Elementsalpatil2No ratings yet
- Bitumen Compound (MSDS)Document4 pagesBitumen Compound (MSDS)meor azrieNo ratings yet
- A 000 989 88 03 11 Servolenkungsöl/ Lenkgetriebeöl MB 236.3Document15 pagesA 000 989 88 03 11 Servolenkungsöl/ Lenkgetriebeöl MB 236.3tuckarytnwNo ratings yet
- Periodic Tales: A Cultural History of the Elements, from Arsenic to ZincFrom EverandPeriodic Tales: A Cultural History of the Elements, from Arsenic to ZincRating: 3.5 out of 5 stars3.5/5 (137)
- The Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactFrom EverandThe Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactRating: 5 out of 5 stars5/5 (5)
- Chemistry: a QuickStudy Laminated Reference GuideFrom EverandChemistry: a QuickStudy Laminated Reference GuideRating: 5 out of 5 stars5/5 (1)
- The Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactFrom EverandThe Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactRating: 5 out of 5 stars5/5 (1)
- ICH Quality Guidelines: An Implementation GuideFrom EverandICH Quality Guidelines: An Implementation GuideAndrew TeasdaleNo ratings yet
- Chemistry for Breakfast: The Amazing Science of Everyday LifeFrom EverandChemistry for Breakfast: The Amazing Science of Everyday LifeRating: 4.5 out of 5 stars4.5/5 (14)
- Essential Chemistry for Formulators of Semisolid and Liquid DosagesFrom EverandEssential Chemistry for Formulators of Semisolid and Liquid DosagesRating: 5 out of 5 stars5/5 (2)
- The Regenerative Grower's Guide to Garden Amendments: Using Locally Sourced Materials to Make Mineral and Biological Extracts and FermentsFrom EverandThe Regenerative Grower's Guide to Garden Amendments: Using Locally Sourced Materials to Make Mineral and Biological Extracts and FermentsRating: 5 out of 5 stars5/5 (3)
- Practical Approaches to Method Validation and Essential Instrument QualificationFrom EverandPractical Approaches to Method Validation and Essential Instrument QualificationNo ratings yet
- The Nature of Drugs Vol. 2: History, Pharmacology, and Social ImpactFrom EverandThe Nature of Drugs Vol. 2: History, Pharmacology, and Social ImpactNo ratings yet
- AP Chemistry Flashcards, Fourth Edition: Up-to-Date Review and PracticeFrom EverandAP Chemistry Flashcards, Fourth Edition: Up-to-Date Review and PracticeNo ratings yet
- Organic Chemistry for Schools: Advanced Level and Senior High SchoolFrom EverandOrganic Chemistry for Schools: Advanced Level and Senior High SchoolNo ratings yet
- Dust Explosion and Fire Prevention Handbook: A Guide to Good Industry PracticesFrom EverandDust Explosion and Fire Prevention Handbook: A Guide to Good Industry PracticesNo ratings yet
- Guidelines for Integrating Process Safety into Engineering ProjectsFrom EverandGuidelines for Integrating Process Safety into Engineering ProjectsNo ratings yet
- The Periodic Table: A Very Short IntroductionFrom EverandThe Periodic Table: A Very Short IntroductionRating: 4.5 out of 5 stars4.5/5 (4)
- Handbook of Formulating Dermal Applications: A Definitive Practical GuideFrom EverandHandbook of Formulating Dermal Applications: A Definitive Practical GuideNo ratings yet
- Lime and Limestone: Chemistry and Technology, Production and UsesFrom EverandLime and Limestone: Chemistry and Technology, Production and UsesRating: 4 out of 5 stars4/5 (1)