You might also like
- Outcome Prediction in CancerFrom EverandOutcome Prediction in CancerAzzam F.G. TaktakNo ratings yet
- Cancer Stem Cell Associated Eight Gene-Based Signature Predicts Clinical Outcomes of Colorectal CancerDocument8 pagesCancer Stem Cell Associated Eight Gene-Based Signature Predicts Clinical Outcomes of Colorectal CancerManoj Kumar pandreNo ratings yet
- Functional Genomics in HCC 2005Document6 pagesFunctional Genomics in HCC 2005johnyap11No ratings yet
- Microarreglos de Carcinoma Hepatocelular.Document14 pagesMicroarreglos de Carcinoma Hepatocelular.planhigion06No ratings yet
- Prediction of Overall Survival Based Upon A New Ferroptosis-Related Gene Signature in Patients With Clear Cell Renal Cell CarcinomaDocument18 pagesPrediction of Overall Survival Based Upon A New Ferroptosis-Related Gene Signature in Patients With Clear Cell Renal Cell Carcinoma肖楚天No ratings yet
- JurnalDocument11 pagesJurnalPuput Indah PratiwiNo ratings yet
- Wo 20 27106Document8 pagesWo 20 27106pentingskripsiNo ratings yet
- A Simple and Reproducible Breast Cancer Prognostic Test: Methodologyarticle Open AccessDocument7 pagesA Simple and Reproducible Breast Cancer Prognostic Test: Methodologyarticle Open AccessMohiuddin JishanNo ratings yet
- HHS Public Access: The Prognostic Landscape of Genes and Infiltrating Immune Cells Across Human CancersDocument28 pagesHHS Public Access: The Prognostic Landscape of Genes and Infiltrating Immune Cells Across Human CancersMohiuddin JishanNo ratings yet
- Cancers PaperDocument17 pagesCancers Paperalpna81No ratings yet
- Identifying Potential Cancer Driver Genes by Genomic Data IntegrationDocument9 pagesIdentifying Potential Cancer Driver Genes by Genomic Data IntegrationAnonymous PKE8zOXNo ratings yet
- Research ArticleDocument16 pagesResearch Articlerezza sesaria yudha perdanaNo ratings yet
- Genomica Del CáncerDocument15 pagesGenomica Del CáncerDaniel PintoNo ratings yet
- 2022 Article 4805Document19 pages2022 Article 4805palvimaneNo ratings yet
- Clinical Value of Consensus Molecular Subtypes in Colorectal CancerDocument14 pagesClinical Value of Consensus Molecular Subtypes in Colorectal CancerOvamelia JulioNo ratings yet
- Identi Fication and Validation of Key Genes With Prognostic Value in Non-Small-Cell Lung Cancer Via Integrated Bioinformatics AnalysisDocument16 pagesIdenti Fication and Validation of Key Genes With Prognostic Value in Non-Small-Cell Lung Cancer Via Integrated Bioinformatics Analysisfee faysal tusarNo ratings yet
- PNAS 2011 Ruiz 1104009108Document6 pagesPNAS 2011 Ruiz 1104009108TomO'MahoneyNo ratings yet
- Xu DKK (2017)Document8 pagesXu DKK (2017)Yoga DinathaNo ratings yet
- CCNB2Document14 pagesCCNB2adamNo ratings yet
- Cytogenetic Profile Predicts Prognosis of Patients With Clear Cell Renal Cell CarcinomaDocument8 pagesCytogenetic Profile Predicts Prognosis of Patients With Clear Cell Renal Cell CarcinomaRivan 21No ratings yet
- Gene Expression Profiling of Renal Cell CarcinomaDocument8 pagesGene Expression Profiling of Renal Cell CarcinomaNepsis ElvețiaNo ratings yet
- Journal of Translational Autoimmunity: Wenbiao Chen, Kefan Bi, Xujun Zhang, Jingjing Jiang, Hongyan DiaoDocument8 pagesJournal of Translational Autoimmunity: Wenbiao Chen, Kefan Bi, Xujun Zhang, Jingjing Jiang, Hongyan DiaoMarinaNo ratings yet
- A Genomic StrategyDocument11 pagesA Genomic StrategyAmirahShalehaNo ratings yet
- ch210005954p PDFDocument5 pagesch210005954p PDFJanNo ratings yet
- 8235.full AACRDocument9 pages8235.full AACRMohammed Khair BashirNo ratings yet
- Cancer 1Document16 pagesCancer 1KlitosNo ratings yet
- Chapter 1 - The Cancer GenomeDocument35 pagesChapter 1 - The Cancer GenomeCynthia LopesNo ratings yet
- Liquid Biopsy Using Ascitic Fluid and Pleural Effusion Supernatants For Genomic Profiling in Gastrointestinal and Lung CancersDocument10 pagesLiquid Biopsy Using Ascitic Fluid and Pleural Effusion Supernatants For Genomic Profiling in Gastrointestinal and Lung CancersCámara de Comercio de CádizNo ratings yet
- 2024 Article 3267Document16 pages2024 Article 3267jabira7113No ratings yet
- Global Computational Alignment of Tumor and Cell Line Transcriptional ProfilesDocument12 pagesGlobal Computational Alignment of Tumor and Cell Line Transcriptional ProfilesRapazito RagazzoNo ratings yet
- Identification of PLAUR-related ceRNA and Immune Prognostic Signature For Kidney Renal Clear Cell CarcinomaDocument18 pagesIdentification of PLAUR-related ceRNA and Immune Prognostic Signature For Kidney Renal Clear Cell Carcinoma肖楚天No ratings yet
- 10.thyroid Cancer TumorigenesisDocument18 pages10.thyroid Cancer TumorigenesisAccounting CV BakerNo ratings yet
- Disease Detection - 2019Document17 pagesDisease Detection - 2019poojaNo ratings yet
- Creatine Phosphokinase Levels in Oral Cancer Patients: Issn 0974-3618 (Print) 0974-360X (Online)Document4 pagesCreatine Phosphokinase Levels in Oral Cancer Patients: Issn 0974-3618 (Print) 0974-360X (Online)Deepak KumarNo ratings yet
- Candidate LncRNA-MiRNA-MRNA Network in PredictingDocument10 pagesCandidate LncRNA-MiRNA-MRNA Network in PredictingBahlibiNo ratings yet
- Prognostic Biomarkers For Survival in Nasopharyngeal Carcinoma - A Systematic Review of The LiteratureDocument14 pagesPrognostic Biomarkers For Survival in Nasopharyngeal Carcinoma - A Systematic Review of The LiteratureWening Dewi HapsariNo ratings yet
- 2008 Annurev - Med. Nanotechnology and CancerDocument18 pages2008 Annurev - Med. Nanotechnology and CancerDenise Gonzalez GomezNo ratings yet
- A Deep Learning Model To Predict RNA-Seq Expression of Tumours From Whole Slide ImagesDocument15 pagesA Deep Learning Model To Predict RNA-Seq Expression of Tumours From Whole Slide ImagesquodicNo ratings yet
- Plasma DNA Integrity Index in Breast Cancer PatientsDocument19 pagesPlasma DNA Integrity Index in Breast Cancer PatientsSalwa Hassan TeamaNo ratings yet
- A New Emerging Picoscale BiotechnologyDocument194 pagesA New Emerging Picoscale BiotechnologyRakesh SharmaNo ratings yet
- Digital Spatial Gene Expression ProfilingDocument16 pagesDigital Spatial Gene Expression Profiling9868838836ankNo ratings yet
- DeepGene - An Advanced Cancer Type Classifier Based On Deep Learning and Somatic Point MutationsDocument14 pagesDeepGene - An Advanced Cancer Type Classifier Based On Deep Learning and Somatic Point MutationstimothyhsesNo ratings yet
- Prediction of Oncogenic Interactions and Cancer-Related Signaling Networks Based On Network TopologyDocument12 pagesPrediction of Oncogenic Interactions and Cancer-Related Signaling Networks Based On Network TopologyFrontiersNo ratings yet
- Bioengineered Three-Dimensional Co-Culture of Cancer Cells and Endothelial Cells A Model System For Dual Analysis of Tumor Growth and AngiogenesisDocument35 pagesBioengineered Three-Dimensional Co-Culture of Cancer Cells and Endothelial Cells A Model System For Dual Analysis of Tumor Growth and Angiogenesisthato69No ratings yet
- Articulo CientificoDocument9 pagesArticulo CientificoHeber Andre Calizaya SánchezNo ratings yet
- Xi, 2021 Tumor Associated Collagen SignaturesDocument15 pagesXi, 2021 Tumor Associated Collagen Signaturesevahendrickx03No ratings yet
- Cancer Genome ProjectDocument6 pagesCancer Genome ProjectDarshil PatelNo ratings yet
- Molecular RCC EmtDocument8 pagesMolecular RCC EmtnadalNo ratings yet
- Convolutional Neural Network Models For Cancer Typ PDFDocument34 pagesConvolutional Neural Network Models For Cancer Typ PDFgisselydszNo ratings yet
- Cancers 12 00037 v2Document24 pagesCancers 12 00037 v2nionNo ratings yet
- Ajcp 134 3 478Document13 pagesAjcp 134 3 478Mita AdrianiNo ratings yet
- 2015 The Cancer Genome AtlasTCGADocument11 pages2015 The Cancer Genome AtlasTCGAJoao da Costa100% (1)
- Circulating DNA As Prognostic Biomarker in Patients With Advanced Hepatocellular Carcinoma, A Translational Exploratory Study From The SORAMIC TrialDocument15 pagesCirculating DNA As Prognostic Biomarker in Patients With Advanced Hepatocellular Carcinoma, A Translational Exploratory Study From The SORAMIC Trialmagendi indra muktiNo ratings yet
- Or 26 6 1539 PDFDocument8 pagesOr 26 6 1539 PDFGloriaaaNo ratings yet
- Breast 2Document7 pagesBreast 2Mina MohammadiNo ratings yet
- Uncovering The Dynamic Effects of DEX Treatment On Lung Ca - 2022 - Computers inDocument10 pagesUncovering The Dynamic Effects of DEX Treatment On Lung Ca - 2022 - Computers indebora eireneNo ratings yet
- Shlien 2010 A Comon Molecular Mechanism Underlies 17p13Document12 pagesShlien 2010 A Comon Molecular Mechanism Underlies 17p13Katherine Lemus SepúlvedaNo ratings yet
- Base-Resolution Stratification of Cancer Mutations Using Functional VariomicsDocument19 pagesBase-Resolution Stratification of Cancer Mutations Using Functional VariomicsPencari IlmuNo ratings yet
- Tumor Marker General Principles and Guidelines (S.sharma 2013)Document8 pagesTumor Marker General Principles and Guidelines (S.sharma 2013)Ria DeviNo ratings yet
- Development of Prognostic Signatures For Intermediate Risk Papillary Thyroid CancerDocument12 pagesDevelopment of Prognostic Signatures For Intermediate Risk Papillary Thyroid Cancernfp.panjaitanNo ratings yet
- Design and Analysis of Cross-Over Trials: Second EditionDocument18 pagesDesign and Analysis of Cross-Over Trials: Second EditionselcukorkmazNo ratings yet
- C0640 BibDocument18 pagesC0640 BibselcukorkmazNo ratings yet
- C0640 ApbDocument4 pagesC0640 ApbselcukorkmazNo ratings yet
- C0640 ApaDocument4 pagesC0640 ApaselcukorkmazNo ratings yet
- Higher-Order Designs For Two Treatments: 2003 CRC Press LLCDocument32 pagesHigher-Order Designs For Two Treatments: 2003 CRC Press LLCselcukorkmazNo ratings yet
- × 2cross-Over TrialDocument107 pages× 2cross-Over TrialselcukorkmazNo ratings yet
- 1.1 What Is A Cross-Over Trial?: × 2 Design. in This Design Each SubjectDocument11 pages1.1 What Is A Cross-Over Trial?: × 2 Design. in This Design Each SubjectselcukorkmazNo ratings yet
- FGS Renal Cell CADocument14 pagesFGS Renal Cell CAselcukorkmazNo ratings yet
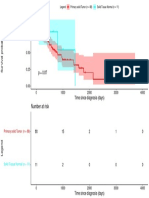
- Number at Risk: Legend Primary Solid Tumor (N 88) Solid Tissue Normal (N 11)Document1 pageNumber at Risk: Legend Primary Solid Tumor (N 88) Solid Tissue Normal (N 11)selcukorkmazNo ratings yet
- FGS Ovarian CADocument12 pagesFGS Ovarian CAselcukorkmazNo ratings yet
- FGS Breast CADocument17 pagesFGS Breast CAselcukorkmazNo ratings yet
- ReadmeDocument17 pagesReadmeJefferson DE Oliveira PaivaNo ratings yet
- Integrated Science Notes Ch10 SenseDocument17 pagesIntegrated Science Notes Ch10 SenseAngel LiNo ratings yet
- 高等学校研究生英语系列教材 综合教程(上)主编熊海虹 教学课件 Unit 1Document129 pages高等学校研究生英语系列教材 综合教程(上)主编熊海虹 教学课件 Unit 1yvaineNo ratings yet
- 2020 International Consensus On Cardiopulmonary Resuscitation and Emergency Cardiovascular Care Science With Treatment Recommendations PDFDocument536 pages2020 International Consensus On Cardiopulmonary Resuscitation and Emergency Cardiovascular Care Science With Treatment Recommendations PDFEduardo Rios DuboisNo ratings yet
- The Behavioral AddictionsDocument238 pagesThe Behavioral AddictionsFarah Farah100% (1)
- Demodex JurnalDocument9 pagesDemodex JurnalRisky PerdanaNo ratings yet
- Acitivity 4Document2 pagesAcitivity 4Elijah Mae MundocNo ratings yet
- PhysioFlow Non Invasive Hemodynamic Monitoring GeneralDocument28 pagesPhysioFlow Non Invasive Hemodynamic Monitoring GeneralPritam HiraNo ratings yet
- 6disorders of Iron Kinetics and Heme MetabolismDocument28 pages6disorders of Iron Kinetics and Heme MetabolismanonacadsNo ratings yet
- BSP Treatment Flow Chart 16 For ScreenDocument2 pagesBSP Treatment Flow Chart 16 For ScreenshahadNo ratings yet
- Article Bladder RetrainingDocument9 pagesArticle Bladder RetrainingRaenette DavidNo ratings yet
- Use of Prebiotic Supplements To Support Gut Microbiome MaintenanceDocument12 pagesUse of Prebiotic Supplements To Support Gut Microbiome MaintenanceIasa FebrianusNo ratings yet
- Lifespan Development Seventh Canadian Edition 7Th Edition Full ChapterDocument41 pagesLifespan Development Seventh Canadian Edition 7Th Edition Full Chapterjamie.gardner403100% (23)
- Basic Life Suppor T: Monalyn B. La-Ao, RN Keverne Colas, RNDocument51 pagesBasic Life Suppor T: Monalyn B. La-Ao, RN Keverne Colas, RNAudi Kyle SaydovenNo ratings yet
- Nonsyndromic Delayed EruptionDocument3 pagesNonsyndromic Delayed EruptionRista LewiyonahNo ratings yet
- Exercise Physiology Lab ReportDocument10 pagesExercise Physiology Lab Reportapi-253305501100% (1)
- Common Nail Disorders J.clindermatol.2013.06.015Document9 pagesCommon Nail Disorders J.clindermatol.2013.06.015Vita BūdvytėNo ratings yet
- Diabetes Blood Sugar Chart Blood Glucose Chart DSMDocument1 pageDiabetes Blood Sugar Chart Blood Glucose Chart DSMBIRD MARKONo ratings yet
- Ya 20.2 Lung Cancer StagingDocument14 pagesYa 20.2 Lung Cancer StagingJuan Francisco Antonio VázquezNo ratings yet
- Human Diseases 1st ChapterDocument10 pagesHuman Diseases 1st Chaptervarun kumarNo ratings yet
- A. Complete Blood Count (CBC)Document6 pagesA. Complete Blood Count (CBC)raul nino MoranNo ratings yet
- The Path To Fitness and Health PE102-Unit1Document24 pagesThe Path To Fitness and Health PE102-Unit1Study ReviewNo ratings yet
- Approach To The Adult With Nausea and Vomiting - UpToDateDocument20 pagesApproach To The Adult With Nausea and Vomiting - UpToDateJolien JobsNo ratings yet
- Test Bank For Mosbys Essentials For Nursing Assistants 4th Edition SorrentinoDocument24 pagesTest Bank For Mosbys Essentials For Nursing Assistants 4th Edition Sorrentinozacharymcleanpdqogjyfeb100% (47)
- Vaginitis: Dr. Zienab Halem Faculty of Pharmacy SCUDocument25 pagesVaginitis: Dr. Zienab Halem Faculty of Pharmacy SCUshona SharupaniNo ratings yet
- Eosinophil Biology and Causes of Eosinophilia - UpToDateDocument35 pagesEosinophil Biology and Causes of Eosinophilia - UpToDateBls PriyaNo ratings yet
- Jethani 2022Document3 pagesJethani 2022febyolaNo ratings yet
- Quick Reference Instructions For Patients: Negative ResultDocument5 pagesQuick Reference Instructions For Patients: Negative ResultShifon StareNo ratings yet
- 2 Prevalence of Sarcopenia in Multi EthnicsDocument10 pages2 Prevalence of Sarcopenia in Multi EthnicsdavidtuanandartuaNo ratings yet
- Perez 2007Document8 pagesPerez 2007SNo ratings yet
- Development and Preliminary Evaluation of PsychomeDocument11 pagesDevelopment and Preliminary Evaluation of Psychomerosa azizahNo ratings yet
- The Age of Magical Overthinking: Notes on Modern IrrationalityFrom EverandThe Age of Magical Overthinking: Notes on Modern IrrationalityRating: 4 out of 5 stars4/5 (23)
- By the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsFrom EverandBy the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsNo ratings yet
- Summary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedFrom EverandSummary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedRating: 5 out of 5 stars5/5 (80)
- Raising Mentally Strong Kids: How to Combine the Power of Neuroscience with Love and Logic to Grow Confident, Kind, Responsible, and Resilient Children and Young AdultsFrom EverandRaising Mentally Strong Kids: How to Combine the Power of Neuroscience with Love and Logic to Grow Confident, Kind, Responsible, and Resilient Children and Young AdultsRating: 5 out of 5 stars5/5 (1)
- Think This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeFrom EverandThink This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeNo ratings yet
- Summary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisFrom EverandSummary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisRating: 4.5 out of 5 stars4.5/5 (42)
- The Obesity Code: Unlocking the Secrets of Weight LossFrom EverandThe Obesity Code: Unlocking the Secrets of Weight LossRating: 4 out of 5 stars4/5 (5)
- Why We Die: The New Science of Aging and the Quest for ImmortalityFrom EverandWhy We Die: The New Science of Aging and the Quest for ImmortalityRating: 4 out of 5 stars4/5 (3)
- The Body Keeps the Score by Bessel Van der Kolk, M.D. - Book Summary: Brain, Mind, and Body in the Healing of TraumaFrom EverandThe Body Keeps the Score by Bessel Van der Kolk, M.D. - Book Summary: Brain, Mind, and Body in the Healing of TraumaRating: 4.5 out of 5 stars4.5/5 (266)
- Mindset by Carol S. Dweck - Book Summary: The New Psychology of SuccessFrom EverandMindset by Carol S. Dweck - Book Summary: The New Psychology of SuccessRating: 4.5 out of 5 stars4.5/5 (328)
- When the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisFrom EverandWhen the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisRating: 3.5 out of 5 stars3.5/5 (2)
- Gut: the new and revised Sunday Times bestsellerFrom EverandGut: the new and revised Sunday Times bestsellerRating: 4 out of 5 stars4/5 (392)
- The Ritual Effect: From Habit to Ritual, Harness the Surprising Power of Everyday ActionsFrom EverandThe Ritual Effect: From Habit to Ritual, Harness the Surprising Power of Everyday ActionsRating: 3.5 out of 5 stars3.5/5 (3)
- Raising Good Humans: A Mindful Guide to Breaking the Cycle of Reactive Parenting and Raising Kind, Confident KidsFrom EverandRaising Good Humans: A Mindful Guide to Breaking the Cycle of Reactive Parenting and Raising Kind, Confident KidsRating: 4.5 out of 5 stars4.5/5 (169)
- The Courage Habit: How to Accept Your Fears, Release the Past, and Live Your Courageous LifeFrom EverandThe Courage Habit: How to Accept Your Fears, Release the Past, and Live Your Courageous LifeRating: 4.5 out of 5 stars4.5/5 (253)
- Dark Psychology & Manipulation: Discover How To Analyze People and Master Human Behaviour Using Emotional Influence Techniques, Body Language Secrets, Covert NLP, Speed Reading, and Hypnosis.From EverandDark Psychology & Manipulation: Discover How To Analyze People and Master Human Behaviour Using Emotional Influence Techniques, Body Language Secrets, Covert NLP, Speed Reading, and Hypnosis.Rating: 4.5 out of 5 stars4.5/5 (110)
- Cult, A Love Story: Ten Years Inside a Canadian Cult and the Subsequent Long Road of RecoveryFrom EverandCult, A Love Story: Ten Years Inside a Canadian Cult and the Subsequent Long Road of RecoveryRating: 4 out of 5 stars4/5 (44)
- 12 Rules for Life by Jordan B. Peterson - Book Summary: An Antidote to ChaosFrom Everand12 Rules for Life by Jordan B. Peterson - Book Summary: An Antidote to ChaosRating: 4.5 out of 5 stars4.5/5 (207)
- How to ADHD: The Ultimate Guide and Strategies for Productivity and Well-BeingFrom EverandHow to ADHD: The Ultimate Guide and Strategies for Productivity and Well-BeingNo ratings yet
- An Autobiography of Trauma: A Healing JourneyFrom EverandAn Autobiography of Trauma: A Healing JourneyRating: 5 out of 5 stars5/5 (2)
- Sleep Stories for Adults: Overcome Insomnia and Find a Peaceful AwakeningFrom EverandSleep Stories for Adults: Overcome Insomnia and Find a Peaceful AwakeningRating: 4 out of 5 stars4/5 (3)
- Outlive: The Science and Art of Longevity by Peter Attia: Key Takeaways, Summary & AnalysisFrom EverandOutlive: The Science and Art of Longevity by Peter Attia: Key Takeaways, Summary & AnalysisRating: 4 out of 5 stars4/5 (1)
- Troubled: A Memoir of Foster Care, Family, and Social ClassFrom EverandTroubled: A Memoir of Foster Care, Family, and Social ClassRating: 4.5 out of 5 stars4.5/5 (26)