You might also like
- Fluctuations and Non-Linear Wave Interactions in Plasmas: International Series in Natural PhilosophyFrom EverandFluctuations and Non-Linear Wave Interactions in Plasmas: International Series in Natural PhilosophyNo ratings yet
- Prentiss - Dissociation of Ligand-Receptor Complexes Using Magnetic TweezersDocument6 pagesPrentiss - Dissociation of Ligand-Receptor Complexes Using Magnetic TweezersChris TranNo ratings yet
- Geometric Effects of Reduction of Dimensionality in Interfacial Reactions'Document5 pagesGeometric Effects of Reduction of Dimensionality in Interfacial Reactions'Dean AstumianNo ratings yet
- J. Am. Chem. Soc. 2001, 123, 1471-1482 (Ag Nano Thiolate)Document12 pagesJ. Am. Chem. Soc. 2001, 123, 1471-1482 (Ag Nano Thiolate)cornelia peñaNo ratings yet
- F Ja00314a008Document1 pageF Ja00314a008Dean AstumianNo ratings yet
- Artigo-1962 - Randolph and Larson - Transient and Steady State Size Distributions in Continuos Mixed Suspension CrystallizersDocument7 pagesArtigo-1962 - Randolph and Larson - Transient and Steady State Size Distributions in Continuos Mixed Suspension CrystallizersAlexandre FonsecaNo ratings yet
- SAW-1-Anal ChemDocument7 pagesSAW-1-Anal ChemsaineelimaNo ratings yet
- Swan Pof 2010Document17 pagesSwan Pof 2010Carlos Josue Amaya CabreraNo ratings yet
- Direct assignment of molecular vibrations in LiZn2Mo3O8 via neutron scatteringDocument24 pagesDirect assignment of molecular vibrations in LiZn2Mo3O8 via neutron scatteringRabbia AminNo ratings yet
- Statistical Theory of The Adsorption of InteractinDocument17 pagesStatistical Theory of The Adsorption of InteractinRamanathan VaradharajanNo ratings yet
- Molecular Dynamics Simulation of Microscopic Droplet Spread On Rough SurfacesDocument8 pagesMolecular Dynamics Simulation of Microscopic Droplet Spread On Rough SurfacesKevin Mathew ThomasNo ratings yet
- 1 s2.0 S0022460X16000407 MainDocument17 pages1 s2.0 S0022460X16000407 MainLexin LINo ratings yet
- Session NN. Underwater Acoustics V: Radiation and ScatteringDocument1 pageSession NN. Underwater Acoustics V: Radiation and ScatteringSowjanya VenigallaNo ratings yet
- Vertical Distribution of Dilute Suspensions in Turbulent PipeDocument9 pagesVertical Distribution of Dilute Suspensions in Turbulent PipePrashant KumarNo ratings yet
- Brownian Motion: A Tool To Determine The Pair Potential Between Colloid ParticlesDocument3 pagesBrownian Motion: A Tool To Determine The Pair Potential Between Colloid ParticlesdeepNo ratings yet
- Contact-Angle Hysteresis SimulationDocument9 pagesContact-Angle Hysteresis SimulationUsha ChevulaNo ratings yet
- Size Distributions in Strongly Coalescing Agitated Liquid-Liquid SystemsDocument4 pagesSize Distributions in Strongly Coalescing Agitated Liquid-Liquid SystemsdonyaNo ratings yet
- Collisionless Energy Transfer in Kinetic Turbulence: Field-Particle Correlations in Fourier SpaceDocument31 pagesCollisionless Energy Transfer in Kinetic Turbulence: Field-Particle Correlations in Fourier SpaceAnindya BiswasNo ratings yet
- Dust Masses, PAH Abundances, and Starlight Intensities in SINGS GalaxiesDocument29 pagesDust Masses, PAH Abundances, and Starlight Intensities in SINGS Galaxies刘丰源No ratings yet
- Cita 5 SdarticleDocument14 pagesCita 5 SdarticleTetrahedro AndrésNo ratings yet
- Detachment DeterminationDocument15 pagesDetachment DeterminationChukwuemeka JosephNo ratings yet
- 2016 - Identifying Factors Which Contribute To The Magnitude of Excess Correlations Between Magnetic Field-Paired Volumes of WaterDocument13 pages2016 - Identifying Factors Which Contribute To The Magnitude of Excess Correlations Between Magnetic Field-Paired Volumes of WaterraduNo ratings yet
- Adams, J. Mann, M. & Ammann, C. (2003) - Proxy Evidence For An El Niño-Like Response To Volcanic ForcingDocument5 pagesAdams, J. Mann, M. & Ammann, C. (2003) - Proxy Evidence For An El Niño-Like Response To Volcanic ForcingNicolas Alberto NuñezNo ratings yet
- The Missing Term in Effective Pair Potentialst: H. J. C. Berendsen, J. R. Grigera, T and T. P. StraatsmaDocument3 pagesThe Missing Term in Effective Pair Potentialst: H. J. C. Berendsen, J. R. Grigera, T and T. P. StraatsmaLuna RamirezNo ratings yet
- Mixing in The Interaction Zone of Two Jets: J, J. J. JDocument10 pagesMixing in The Interaction Zone of Two Jets: J, J. J. JGustavo Gabriel JimenezNo ratings yet
- Numerical Simulation of Particle Deposition in Turbulent Boundary LayersDocument14 pagesNumerical Simulation of Particle Deposition in Turbulent Boundary LayerspdfsinghNo ratings yet
- Spatial Cross-Correlation Method Enhances Surface Wave DispersionDocument10 pagesSpatial Cross-Correlation Method Enhances Surface Wave DispersionHoracio Ramos SaldañaNo ratings yet
- On Transition To Cellularity in Expanding Spherical Ames: G.Jomaas, C. K. L A W J.K.BechtoldDocument26 pagesOn Transition To Cellularity in Expanding Spherical Ames: G.Jomaas, C. K. L A W J.K.Bechtoldigor VladimirovichNo ratings yet
- Meta-Analysis of The Space Ight and Microgravity Response of The Arabidopsis Plant TranscriptomeDocument15 pagesMeta-Analysis of The Space Ight and Microgravity Response of The Arabidopsis Plant Transcriptomevse.superyou.worbyNo ratings yet
- Software Dockking 1Document12 pagesSoftware Dockking 1Ira WiastutiNo ratings yet
- Tortuosity in Porous Media: A Critical Review: Behzad GhanbarianDocument18 pagesTortuosity in Porous Media: A Critical Review: Behzad Ghanbarianayman jummaNo ratings yet
- SSAdNDP Tool Deciphers Multi-Center Bonding in Periodic SystemsDocument8 pagesSSAdNDP Tool Deciphers Multi-Center Bonding in Periodic SystemsA Fernando CGNo ratings yet
- Charge Transfer Conjugated Polymers Science 2009Document3 pagesCharge Transfer Conjugated Polymers Science 2009PengHanNo ratings yet
- Discrimination of Adsorption Kinetic Models For The Description of Hydrocarbon Adsorption in Activated CarbonDocument13 pagesDiscrimination of Adsorption Kinetic Models For The Description of Hydrocarbon Adsorption in Activated CarbonAries ArcegaNo ratings yet
- Geostatistical Analysis of An Experimental StratigraphyDocument20 pagesGeostatistical Analysis of An Experimental StratigraphyDanny SubramaniamNo ratings yet
- 07-1997-Discovery of A Reactive AzeotropeDocument3 pages07-1997-Discovery of A Reactive Azeotropeehsan zeraatkarNo ratings yet
- Shubin 1994Document8 pagesShubin 1994yurdanurturkerNo ratings yet
- Allometric Scaling of Plant Energetics and Population DensityDocument3 pagesAllometric Scaling of Plant Energetics and Population DensityRomina MercadanteNo ratings yet
- Update on electromagnetic basis for inertia, gravitation and particle propertiesDocument11 pagesUpdate on electromagnetic basis for inertia, gravitation and particle propertiesMilan StepanovNo ratings yet
- A Comparison and Test of Various Site-Response Estimation Techniques, Including Three That Are Not Reference-Site DependentDocument18 pagesA Comparison and Test of Various Site-Response Estimation Techniques, Including Three That Are Not Reference-Site Dependentnicolas españaNo ratings yet
- Theoretical Aspects of A Discrete-Binding Approach in Quartz-Crystal Microbalance Acoustic BiosensingDocument22 pagesTheoretical Aspects of A Discrete-Binding Approach in Quartz-Crystal Microbalance Acoustic BiosensingpsephisNo ratings yet
- Vidali 1991Document47 pagesVidali 1991Suerfu BurkhantNo ratings yet
- A Two-Dimensional Study of The Rupture of Funicular LiquidbridgesDocument16 pagesA Two-Dimensional Study of The Rupture of Funicular Liquidbridgesohar1963No ratings yet
- P W Anderson 53736Document14 pagesP W Anderson 53736mattia paganelliNo ratings yet
- Variations of Ionization Potential and Electron Affinity As A Function of Surface Orientation: The Case of Orthorhombic SnsDocument5 pagesVariations of Ionization Potential and Electron Affinity As A Function of Surface Orientation: The Case of Orthorhombic SnsFELIPE ANDRÉS OLIVARES CODOCEONo ratings yet
- American Association For The Advancement of ScienceDocument5 pagesAmerican Association For The Advancement of ScienceNamu PatelNo ratings yet
- H.L. Stormer and D.C. Tsui - The Fractional Quantum Hall EffectDocument2 pagesH.L. Stormer and D.C. Tsui - The Fractional Quantum Hall EffectGreamxxNo ratings yet
- Use of Ab Initio Quantum Molecular Simil PDFDocument7 pagesUse of Ab Initio Quantum Molecular Simil PDFRasyidNo ratings yet
- First Combined Studies On Lorentz Invariance Violation From Observations of Astrophysical SourcesDocument8 pagesFirst Combined Studies On Lorentz Invariance Violation From Observations of Astrophysical Sourcespicard82No ratings yet
- Larose Etal Geophysics2006Document11 pagesLarose Etal Geophysics2006zelenkoNo ratings yet
- The General Problem of Elastic Wave Propagation in Multilayered Anisotropic MediaDocument11 pagesThe General Problem of Elastic Wave Propagation in Multilayered Anisotropic MediaPyrsos CandlesNo ratings yet
- Changes in Charge Density Vs Changes in Formal Oxidation States: The Case of SN Halide Perovskites and Their Ordered Vacancy AnaloguesDocument36 pagesChanges in Charge Density Vs Changes in Formal Oxidation States: The Case of SN Halide Perovskites and Their Ordered Vacancy AnaloguesikoutsNo ratings yet
- Greenhoe Et Al-2016-Journal of Polymer Science Part B - Polymer Physics JournalwebsiteDocument7 pagesGreenhoe Et Al-2016-Journal of Polymer Science Part B - Polymer Physics JournalwebsiteMoad BarbariNo ratings yet
- Microdosimetry of Internally Deposited Radionuclides: Int. J. Appl. Radiat. IsotDocument13 pagesMicrodosimetry of Internally Deposited Radionuclides: Int. J. Appl. Radiat. IsotHalaj AzadNo ratings yet
- Pras Her 1973Document4 pagesPras Her 1973zfreddyzzzNo ratings yet
- s0927 77572900438 0 PDFDocument15 pagess0927 77572900438 0 PDFRyan RamalhoNo ratings yet
- Arnett 1980Document11 pagesArnett 1980Muhamad Zunanda AjiNo ratings yet
- 96 LANG Huibers ShahDocument9 pages96 LANG Huibers ShahCarla M.BizamaNo ratings yet
- J100617a012 PDFDocument11 pagesJ100617a012 PDFChristian PaterninaNo ratings yet
- Discussion of Dynamic Response of Arbitrarily Shaped Foundations Experimental VerificationDocument3 pagesDiscussion of Dynamic Response of Arbitrarily Shaped Foundations Experimental VerificationCarlosAlbertoBarriosnuevosPelaezNo ratings yet
- Astm D 3363-05Document3 pagesAstm D 3363-05briham100% (1)
- Writing A Letter of Apology and CompensationDocument10 pagesWriting A Letter of Apology and CompensationEco ReyNo ratings yet
- LearnEnglish Listening B1 A Team Meeting About DiversityDocument4 pagesLearnEnglish Listening B1 A Team Meeting About Diversitykorespondesi b.inggrisNo ratings yet
- Astm D522 PDFDocument4 pagesAstm D522 PDFelangorenga0% (1)
- 4.3.2 Circular MotionDocument8 pages4.3.2 Circular MotionMozammel AnowarNo ratings yet
- The Consep AcaciaDocument7 pagesThe Consep AcaciaPrincess Ruwarashe Courteney MunyoroNo ratings yet
- Rubiag 13Document1 pageRubiag 13ChérubinNo ratings yet
- Question 926614Document7 pagesQuestion 926614davulurusurekhaNo ratings yet
- Experiment 6 Lab ReportDocument21 pagesExperiment 6 Lab ReportmarkkkkkNo ratings yet
- Widmanstätten StructuresDocument3 pagesWidmanstätten StructuresdantegimenezNo ratings yet
- Shieldgrout CG1Document3 pagesShieldgrout CG1malak hindiNo ratings yet
- Masterprotect 1812 TdsDocument2 pagesMasterprotect 1812 TdsIko SchyterNo ratings yet
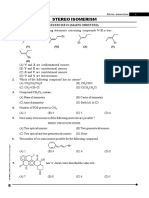
- Stereo Isomerism - (Eng)Document32 pagesStereo Isomerism - (Eng)Atharva WatekarNo ratings yet
- Armix Hyyecrete Sp200: - Forhighgradeandmicro-SilicaconcreteDocument3 pagesArmix Hyyecrete Sp200: - Forhighgradeandmicro-Silicaconcreteraviteja036No ratings yet
- 01 To UTBK Full Version (SAINTEK) - Bahasa InggrisDocument8 pages01 To UTBK Full Version (SAINTEK) - Bahasa Inggrissepia wulandariNo ratings yet
- WLP and DLP3 Aug. 30 Sep. 3 Earths Internal StructureDocument4 pagesWLP and DLP3 Aug. 30 Sep. 3 Earths Internal StructureMariel LolincoNo ratings yet
- Deflection of Beams Double Integration MethodDocument4 pagesDeflection of Beams Double Integration MethodJoeron Caezar Del Rosario (Joe)No ratings yet
- Chapter 38 - Photons and Matter WavesDocument12 pagesChapter 38 - Photons and Matter WavesVV Cephei100% (1)
- 230 Sp18 PREP5 Blank PDFDocument24 pages230 Sp18 PREP5 Blank PDFKhang HuynhNo ratings yet
- Localized Indigenized Instructional Materials in Science 9Document6 pagesLocalized Indigenized Instructional Materials in Science 9joyce kris AlmacinNo ratings yet
- Isomerization of Aldoximes to Amides Under Neutral ConditionsDocument5 pagesIsomerization of Aldoximes to Amides Under Neutral ConditionsDavideGiacomelliNo ratings yet
- IECEx BAS 14.0097X 003Document11 pagesIECEx BAS 14.0097X 003Marcos SiqueiraNo ratings yet
- Clariant Chemical Taiwan Roadshow Highlights Key Issues in Leather DyeingDocument74 pagesClariant Chemical Taiwan Roadshow Highlights Key Issues in Leather Dyeing高文恩No ratings yet
- Entropy Changes in A Chemical ReactionDocument3 pagesEntropy Changes in A Chemical ReactionSudhaNo ratings yet
- Exposición - Grupo 5Document20 pagesExposición - Grupo 5Santiago DíazNo ratings yet
- Concrete Mix DesignDocument21 pagesConcrete Mix DesignfaheemqcNo ratings yet
- Agilent HPLC and UHPLC Application HighlightsDocument159 pagesAgilent HPLC and UHPLC Application HighlightsHeather Fleming100% (1)
- Paribhashik ShabdawaliDocument5 pagesParibhashik Shabdawalisgangwar2005sg100% (1)
- Cambridge IGCSE™: Chemistry 0620/62 May/June 2022Document7 pagesCambridge IGCSE™: Chemistry 0620/62 May/June 2022aayan rahmanNo ratings yet
- The Ncuk International Foundation Year IFYCH002 Chemistry End of Semester 1 Test 2018-19Document12 pagesThe Ncuk International Foundation Year IFYCH002 Chemistry End of Semester 1 Test 2018-19MfanafuthiNo ratings yet
- Pondicherry University B.Sc. Physics Regulations & SyllabusDocument39 pagesPondicherry University B.Sc. Physics Regulations & SyllabusenochrajeshNo ratings yet
- Waterex ThickenersDocument2 pagesWaterex ThickenerssaravananNo ratings yet
- Isometric Gradient Social Media Strategy by SlidesgoDocument46 pagesIsometric Gradient Social Media Strategy by SlidesgoEwerton MazoniNo ratings yet
- Iesc 103Document12 pagesIesc 103Debasish DeyNo ratings yet