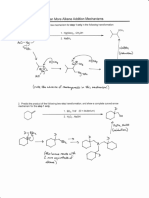
You might also like
- The Total Synthesis of Natural ProductsFrom EverandThe Total Synthesis of Natural ProductsJohn ApSimonNo ratings yet
- Novel Synthesis of Benzofuran - and Indol-2-Yl-Methanamine Derivatives PDFDocument6 pagesNovel Synthesis of Benzofuran - and Indol-2-Yl-Methanamine Derivatives PDFMiguelAlejandroMantaChavezNo ratings yet
- Carbohydrate Chemistry—8: Plenary Lectures Presented at the Eighth International Symposium on Carbohydrate Chemistry, Kyoto, Japan 16 - 20 August 1976From EverandCarbohydrate Chemistry—8: Plenary Lectures Presented at the Eighth International Symposium on Carbohydrate Chemistry, Kyoto, Japan 16 - 20 August 1976K. OnoderaNo ratings yet
- Vogel Natural and Non-Natural Prenylated ChalconesDocument8 pagesVogel Natural and Non-Natural Prenylated Chalconespjalat bpibNo ratings yet
- Synthesis of Some Novel Isoxazoline From Sec 3-Amino Pyridazine Chalcones and Their Antimicrobial StudiesDocument8 pagesSynthesis of Some Novel Isoxazoline From Sec 3-Amino Pyridazine Chalcones and Their Antimicrobial StudiesMJBAS JournalNo ratings yet
- Stereoselective Total Synthesis (+) - Artemisinin, The Antimalarial ConstituentDocument6 pagesStereoselective Total Synthesis (+) - Artemisinin, The Antimalarial ConstituentTom ChanNo ratings yet
- Total Synthesis and Antiviral Activity of Enantioenriched (+) - Deoxytylo-PhorinineDocument5 pagesTotal Synthesis and Antiviral Activity of Enantioenriched (+) - Deoxytylo-PhorinineabcdefNo ratings yet
- Erythroxylon Coca : Biosynthesis of Cocaine and Cuscohygrine From (1 - 4C) ACETATE and (4-3H) Phenylalanine INDocument6 pagesErythroxylon Coca : Biosynthesis of Cocaine and Cuscohygrine From (1 - 4C) ACETATE and (4-3H) Phenylalanine INDaniel ChamorroNo ratings yet
- s0957 4166 (00) 80369 9 PDFDocument6 pagess0957 4166 (00) 80369 9 PDFMike RohrichNo ratings yet
- 'H NMR Green: and 13C Assignments of Some Tea PolyphenolsDocument4 pages'H NMR Green: and 13C Assignments of Some Tea PolyphenolsPlant VietNo ratings yet
- Synthesis of Multisubstituted Furans, Pyrroles, and Thiophenes Via YnolatesDocument4 pagesSynthesis of Multisubstituted Furans, Pyrroles, and Thiophenes Via YnolatesSaurav PaulNo ratings yet
- El Khoury 2010Document33 pagesEl Khoury 2010Ngọc Phương Vy NguyễnNo ratings yet
- RtyDocument22 pagesRtyWalid Ebid ElgammalNo ratings yet
- Cofactors MainDocument16 pagesCofactors MainmigenyasuyoshiNo ratings yet
- Alpha Spinasterol in The Roots of Impatiens Glandulifera and Its Effects On The Viability of Human CellsDocument9 pagesAlpha Spinasterol in The Roots of Impatiens Glandulifera and Its Effects On The Viability of Human Cellsrrbd33No ratings yet
- Reação Butil LitioDocument7 pagesReação Butil LitioArtur RamonNo ratings yet
- Chemical Communications,: Organic Derivatives of A Cobalt (Macrocylic ComplexDocument2 pagesChemical Communications,: Organic Derivatives of A Cobalt (Macrocylic ComplexRoz RozadoNo ratings yet
- Bioorganic & Medicinal Chemistry LettersDocument4 pagesBioorganic & Medicinal Chemistry LettersmiltonNo ratings yet
- Iranian Chemical Society: Department of Chemistry, College of Science, Shahid Chamran University, Ahvaz, IranDocument7 pagesIranian Chemical Society: Department of Chemistry, College of Science, Shahid Chamran University, Ahvaz, IranjocyeoNo ratings yet
- Reactions With Heterocyclic - Enaminoesters: A Novel Synthesis of 2-Amino-3-Ethoxycarbonyl - (4H) - PyransDocument5 pagesReactions With Heterocyclic - Enaminoesters: A Novel Synthesis of 2-Amino-3-Ethoxycarbonyl - (4H) - PyransWalid EbaiedNo ratings yet
- Isoeugenol Ester Derivatives As Future Potential DrugDocument6 pagesIsoeugenol Ester Derivatives As Future Potential DrugIJRASETPublicationsNo ratings yet
- Synthesis and Antimicrobial Evaluation of Some Isoxazolidine DerivativesDocument9 pagesSynthesis and Antimicrobial Evaluation of Some Isoxazolidine Derivativesمحمد بلحوتNo ratings yet
- 28sici 291099 0518 28199702 2935 3A3 3C575 3A 3aaid Pola23 3e3.0.co 3B2 KDocument5 pages28sici 291099 0518 28199702 2935 3A3 3C575 3A 3aaid Pola23 3e3.0.co 3B2 KLata DeshmukhNo ratings yet
- Articulo 1Document5 pagesArticulo 1Melissa Silva MarinNo ratings yet
- A New Sesquiterpene Lactone From The Roots of Saussurea Lappa Structure-Anticancer Activity StudyDocument3 pagesA New Sesquiterpene Lactone From The Roots of Saussurea Lappa Structure-Anticancer Activity StudyTuan NguyenNo ratings yet
- Research Article: Study of Catalytic Hydrogenation and Methanol Addition ToDocument4 pagesResearch Article: Study of Catalytic Hydrogenation and Methanol Addition ToAni Yunita SariNo ratings yet
- Adducts PaperDocument5 pagesAdducts PaperFrequency!No ratings yet
- One-Pot Highly Enantioselective Catalyti PDFDocument4 pagesOne-Pot Highly Enantioselective Catalyti PDFDaniel HernándezNo ratings yet
- A Review On Synthesis and Medicinal Importance of Thiophene: Raghav Mishra, Pramod Kumar SharmaDocument14 pagesA Review On Synthesis and Medicinal Importance of Thiophene: Raghav Mishra, Pramod Kumar SharmaTanmay WalunjNo ratings yet
- 2010 TLDocument6 pages2010 TLSudha PriyaNo ratings yet
- ) Cychze: Synthesis of Benzo-Fused, 7,5-And 7,6-Fused Azepinones As Conformationally Restricted Dipeptide MimeticsDocument4 pages) Cychze: Synthesis of Benzo-Fused, 7,5-And 7,6-Fused Azepinones As Conformationally Restricted Dipeptide MimeticsAngie Melendez MendezNo ratings yet
- Venkat Rag A Van 2009Document6 pagesVenkat Rag A Van 2009Irene ValdiviesoNo ratings yet
- Delayen A. Heterocycle 2005, 65 - 1673Document6 pagesDelayen A. Heterocycle 2005, 65 - 1673nileshshitole619No ratings yet
- 01SL1731Document4 pages01SL1731SOMMENNo ratings yet
- Synthesis and Analgesic Evaluation of Some 5 - (B - (10-Phenothiazinyl) Ethyl) - 1 - (Acyl) - 1,2,3,4-TetrazolesDocument7 pagesSynthesis and Analgesic Evaluation of Some 5 - (B - (10-Phenothiazinyl) Ethyl) - 1 - (Acyl) - 1,2,3,4-TetrazolesWalid EbaiedNo ratings yet
- 102 CH 15A - Aldehyde and Ketone Structures and NamesDocument16 pages102 CH 15A - Aldehyde and Ketone Structures and NamesGracie OlsonNo ratings yet
- Acetogenins From Annona Glabra SeedsDocument6 pagesAcetogenins From Annona Glabra SeedsQuyen DoNo ratings yet
- Preparation of 2-Alkylidene Oxetanes: An Investigation of The Paterno-Biichi Reaction Between Aliphatic Aldehydes and AllenesDocument4 pagesPreparation of 2-Alkylidene Oxetanes: An Investigation of The Paterno-Biichi Reaction Between Aliphatic Aldehydes and AllenesSaurav PaulNo ratings yet
- Isolation of Campesteryl Ferulate and Epi Campesteryl Ferulate Two Components of Oryzanol From Rice BranDocument4 pagesIsolation of Campesteryl Ferulate and Epi Campesteryl Ferulate Two Components of Oryzanol From Rice Branthiên vũ hoàngNo ratings yet
- An Efficient Synthesis of Benzochromeno-Pyrazoles: Hossein Dianat, Alireza Nazif, Saeid SalimiDocument3 pagesAn Efficient Synthesis of Benzochromeno-Pyrazoles: Hossein Dianat, Alireza Nazif, Saeid SalimierpublicationNo ratings yet
- A Highly Active Catalyst For The Hydrogenation of Amides To Alcoholsand AminesDocument4 pagesA Highly Active Catalyst For The Hydrogenation of Amides To Alcoholsand Aminesxuyijing2007comNo ratings yet
- Tetrahedron Letters: Graziano Baccolini, Carla Boga, Camilla Delpivo, Gabriele MichelettiDocument5 pagesTetrahedron Letters: Graziano Baccolini, Carla Boga, Camilla Delpivo, Gabriele MichelettiRiyadh RayhandhiaNo ratings yet
- Saraei2015 Article SynthesisOfNovelTrans-stilbeneDocument6 pagesSaraei2015 Article SynthesisOfNovelTrans-stilbeneJuan MartínezNo ratings yet
- Efficient Synthesis of Benzo...Document7 pagesEfficient Synthesis of Benzo...Douglas DíazNo ratings yet
- Synthesis and Evaluation of Some Variants of The Nefkens' ReagentDocument3 pagesSynthesis and Evaluation of Some Variants of The Nefkens' Reagentlost6taNo ratings yet
- Synthesis of 1,2,3,4,5-Pentasubstituted Symmetrical PyrrolesDocument5 pagesSynthesis of 1,2,3,4,5-Pentasubstituted Symmetrical PyrrolesthirukkannansastikaNo ratings yet
- Characterization of P-Menthanols and P-Menthanyl Acetates: David H. DDocument7 pagesCharacterization of P-Menthanols and P-Menthanyl Acetates: David H. DРусланNo ratings yet
- PIIS0021925817390488Document5 pagesPIIS0021925817390488ArturoNo ratings yet
- Synthesis of New Paratoluene Sulphonamide Derivatives of Amino Acids and Their Anti Bacterial ActivitiesDocument4 pagesSynthesis of New Paratoluene Sulphonamide Derivatives of Amino Acids and Their Anti Bacterial ActivitiesDarian HerascuNo ratings yet
- 6171 Synthesis of N Isobutylnoroxymorphone From Naltrexone by A Selective Cyclopropane Ring Opening Reactionb77b PDFDocument4 pages6171 Synthesis of N Isobutylnoroxymorphone From Naltrexone by A Selective Cyclopropane Ring Opening Reactionb77b PDFlumik1234No ratings yet
- European Journal of Medicinal Chemistry: Channamata Shankara Naveena, Poojary Boja, Nalilu Sucheta KumariDocument12 pagesEuropean Journal of Medicinal Chemistry: Channamata Shankara Naveena, Poojary Boja, Nalilu Sucheta KumariWalid Ebid ElgammalNo ratings yet
- Cukurovali 2006Document7 pagesCukurovali 2006jipir64332No ratings yet
- European Journal of Medicinal Chemistry: 1 Sahar Mahmoud Abou-Seri, Khaled Abouzid, Dalal A. Abou El EllaDocument12 pagesEuropean Journal of Medicinal Chemistry: 1 Sahar Mahmoud Abou-Seri, Khaled Abouzid, Dalal A. Abou El EllaWalid EbaiedNo ratings yet
- Umihara Et Al-2017-Chemistry - A European JournalDocument3 pagesUmihara Et Al-2017-Chemistry - A European JournalNathalia MojicaNo ratings yet
- Biskra Sayad RayeneDocument1 pageBiskra Sayad Rayenerayene sayadNo ratings yet
- (Elearnica - Ir) - Towards The Total Synthesis of Tashironin Related Allo-Cedrane Natural ProdDocument4 pages(Elearnica - Ir) - Towards The Total Synthesis of Tashironin Related Allo-Cedrane Natural ProdMohamadMostafaviNo ratings yet
- LC-MS Compatible Stability Indicating RP-UPLC Method For The Estimation of Ester Prodrug of Mycophenolic Acid in Injection FormulationDocument10 pagesLC-MS Compatible Stability Indicating RP-UPLC Method For The Estimation of Ester Prodrug of Mycophenolic Acid in Injection FormulationRatnakaram Venkata NadhNo ratings yet
- Sandu Lenko 2016Document6 pagesSandu Lenko 2016alfina ameliaNo ratings yet
- 1984 - WadsworthDkk - Enamines From Iodine Oxidation of Trialkylamines. 1. Electrophilic Capture by Cationic Heterocyclic RingsDocument6 pages1984 - WadsworthDkk - Enamines From Iodine Oxidation of Trialkylamines. 1. Electrophilic Capture by Cationic Heterocyclic RingsUAS TekimNo ratings yet
- Selected Medicinal PlantsDocument10 pagesSelected Medicinal PlantsNegar RzdNo ratings yet
- Chemistry: AN JournalDocument14 pagesChemistry: AN JournalQuốc NguyễnNo ratings yet
- Chemistry 6941, Fall 2007 Synthesis Problems I - Key Dr. Peter NorrisDocument9 pagesChemistry 6941, Fall 2007 Synthesis Problems I - Key Dr. Peter NorrisQuốc NguyễnNo ratings yet
- Kummer 2005Document3 pagesKummer 2005Quốc NguyễnNo ratings yet
- Aluminium Aquo SpeciationDocument1 pageAluminium Aquo SpeciationQuốc NguyễnNo ratings yet
- CHM B44Y Test 3Document15 pagesCHM B44Y Test 3Quốc NguyễnNo ratings yet
- Part One: Mirrors Part Two: Synthesis of Camphor: by Robert C. KentDocument20 pagesPart One: Mirrors Part Two: Synthesis of Camphor: by Robert C. KentQuốc NguyễnNo ratings yet
- 43 Austrian Chemistry Olympiad National Competition Theoretical Tasks 2017-05-25Document35 pages43 Austrian Chemistry Olympiad National Competition Theoretical Tasks 2017-05-25Quốc NguyễnNo ratings yet
- INChO2017 QuestionDocument30 pagesINChO2017 QuestionTheStupidityOfPplNo ratings yet
- Problems 2nd Round Schema1Document1 pageProblems 2nd Round Schema1Quốc NguyễnNo ratings yet
- Dokumen - Pub A Problem Book in Chemistry For Iit Jee 1500 Selected Questions 9789312147382Document717 pagesDokumen - Pub A Problem Book in Chemistry For Iit Jee 1500 Selected Questions 9789312147382Quốc NguyễnNo ratings yet
- Reviews of Workbook For Organic ChemistrDocument2 pagesReviews of Workbook For Organic ChemistrQuốc NguyễnNo ratings yet
- Ozone Cyanide Removal July 29Document8 pagesOzone Cyanide Removal July 29Quốc NguyễnNo ratings yet
- 47 Ukrainian Chemistry Olympiad: Final National CompetitionDocument39 pages47 Ukrainian Chemistry Olympiad: Final National CompetitionQuốc NguyễnNo ratings yet
- Preparatory Problems Icho 2019Document200 pagesPreparatory Problems Icho 2019Quốc NguyễnNo ratings yet
- A B C D Answer: B, A, D, C: O O BR 1 Eq Br2 Febr3Document3 pagesA B C D Answer: B, A, D, C: O O BR 1 Eq Br2 Febr3Quốc NguyễnNo ratings yet
- Week 7 Prac Prob SolnsDocument9 pagesWeek 7 Prac Prob SolnsQuốc NguyễnNo ratings yet
- Alcohol Protecting Groups: OTHP/OMOM Protecting GroupDocument7 pagesAlcohol Protecting Groups: OTHP/OMOM Protecting GroupQuốc NguyễnNo ratings yet
- Alcohol Protecting Groups: OTHP/OMOM Protecting GroupDocument7 pagesAlcohol Protecting Groups: OTHP/OMOM Protecting GroupQuốc NguyễnNo ratings yet
- Problems 2nd Round Schema1Document1 pageProblems 2nd Round Schema1Quốc NguyễnNo ratings yet
- Questions and Answers - Elimination ReactionDocument17 pagesQuestions and Answers - Elimination ReactionQuốc NguyễnNo ratings yet
- Marine GlycosidesDocument266 pagesMarine GlycosidesQuốc NguyễnNo ratings yet
- Imcho2020s.en 2Document15 pagesImcho2020s.en 2Quốc NguyễnNo ratings yet
- Problem 1 (Author Khvalyuk V.N.) : 54th International Mendeleev Olympiad, 2020 1 Theoretical Tour SolutionsDocument15 pagesProblem 1 (Author Khvalyuk V.N.) : 54th International Mendeleev Olympiad, 2020 1 Theoretical Tour SolutionsQuốc NguyễnNo ratings yet
- 1 3 Cyclohexanedione and Its DerivativesDocument55 pages1 3 Cyclohexanedione and Its DerivativesQuốc NguyễnNo ratings yet
- 2004 RD 1 Answers tcm18-190747Document8 pages2004 RD 1 Answers tcm18-190747LouiseflemingNo ratings yet
- Practice Exam 3ADocument10 pagesPractice Exam 3AQuốc NguyễnNo ratings yet
- 2003 RD 1 Answers tcm18-190749Document6 pages2003 RD 1 Answers tcm18-190749LouiseflemingNo ratings yet
- Chapter 4 Aldehydes and Ketones ChapterDocument9 pagesChapter 4 Aldehydes and Ketones ChapterQuốc NguyễnNo ratings yet
- Chapter 4 Aldehydes and Ketones ChapterDocument9 pagesChapter 4 Aldehydes and Ketones ChapterQuốc NguyễnNo ratings yet
- DGFGHDocument4 pagesDGFGHKaur AnjandeepNo ratings yet
- Synthesis of Phenytoin From Benzil and Urea: RequirementsDocument24 pagesSynthesis of Phenytoin From Benzil and Urea: RequirementsTejas ShindeNo ratings yet
- 1998v46no1p206 210Document5 pages1998v46no1p206 210Glédyson FerreiraNo ratings yet
- Svenningsen 2015 NC c2015Document19 pagesSvenningsen 2015 NC c2015AzizNo ratings yet
- Evaluating Impurities in Drugs - Part III of IIIDocument8 pagesEvaluating Impurities in Drugs - Part III of IIIwindli2014No ratings yet
- HPTLCDocument28 pagesHPTLCVshn VardhnNo ratings yet
- FST MCQS 5 With Answers Key For PFADocument42 pagesFST MCQS 5 With Answers Key For PFAIqra MustafaNo ratings yet
- Dr. Sarita Khaturia CVDocument13 pagesDr. Sarita Khaturia CVGloria Ryan KhatriNo ratings yet
- Datura Metel Alkaloid ContentDocument14 pagesDatura Metel Alkaloid ContentpomodoroNo ratings yet
- 1 Pharmaceutical Analysis 2 Comprehensive ReviewerDocument8 pages1 Pharmaceutical Analysis 2 Comprehensive Reviewertahera didaagunNo ratings yet
- Chemistry3 96) PDocument43 pagesChemistry3 96) PShlok ParekhNo ratings yet
- Caffeine Extraction 1 PDFDocument25 pagesCaffeine Extraction 1 PDFShanay ShahNo ratings yet
- Glyphosate RAR 08 Volume 3CA-CP B-6 2013-12-18 SanDocument947 pagesGlyphosate RAR 08 Volume 3CA-CP B-6 2013-12-18 SanHungry4PesticidesNo ratings yet
- Experiment 8 TLCDocument2 pagesExperiment 8 TLCMsfaeza HanafiNo ratings yet
- Chen2017 - Morphological and Chemical Variation of Stemona Tuberosa From MEDICINAL USESDocument8 pagesChen2017 - Morphological and Chemical Variation of Stemona Tuberosa From MEDICINAL USESanascopelNo ratings yet
- 3.1 Pre-Laboratory For The Chromatographic Separation of Amino Acids - GUMAHINDocument3 pages3.1 Pre-Laboratory For The Chromatographic Separation of Amino Acids - GUMAHINJoan Erika GumahinNo ratings yet
- Note Thin-Layer Chromatography of Benzoylecgonine: A Rapid Qualitative Method For Confirming The EMIT Urine Cocaine Metabolite AssaysDocument7 pagesNote Thin-Layer Chromatography of Benzoylecgonine: A Rapid Qualitative Method For Confirming The EMIT Urine Cocaine Metabolite AssaysPara CelseNo ratings yet
- Lbymatc Lab Handouts Ay 2014-2015Document31 pagesLbymatc Lab Handouts Ay 2014-2015Stefanie EstilloreNo ratings yet
- Isolation and Antibacterial Activity of Terpenoid From: Available Online atDocument4 pagesIsolation and Antibacterial Activity of Terpenoid From: Available Online atDeni PratamaNo ratings yet
- D 03 Man Aspirin Syn AnalysisDocument12 pagesD 03 Man Aspirin Syn AnalysisWahyu RedfieldNo ratings yet
- Andrade Et Al. 2006Document5 pagesAndrade Et Al. 2006Tatiana AndradeNo ratings yet
- Experiment 2 TLCDocument6 pagesExperiment 2 TLCAnonymous 75TDy2yNo ratings yet
- Preliminary Pharmacognosy Evaluation of The Yashtimadhu Glycyrrhiza Glebra (Linn.)Document5 pagesPreliminary Pharmacognosy Evaluation of The Yashtimadhu Glycyrrhiza Glebra (Linn.)International Journal of Innovative Science and Research TechnologyNo ratings yet
- Research Article: Validation of A UV Spectrometric Method For The Assay of Tolfenamic Acid in Organic SolventsDocument19 pagesResearch Article: Validation of A UV Spectrometric Method For The Assay of Tolfenamic Acid in Organic SolventsAni Yunita SariNo ratings yet
- Instruction Manual: Automatic TLC Sampler 4Document33 pagesInstruction Manual: Automatic TLC Sampler 4amoricz1No ratings yet
- Prednisolone Acetate: Prednisoloni AcetasDocument3 pagesPrednisolone Acetate: Prednisoloni AcetasMarcosNo ratings yet
- Analysis of Analgesic Tablets Using Thin-Layer ChromatographyDocument6 pagesAnalysis of Analgesic Tablets Using Thin-Layer ChromatographyJoleigh Delos ReyesNo ratings yet
- Monographs of DrugsDocument523 pagesMonographs of DrugsVishal ParmarNo ratings yet
- Chem 160.1 Exer 9 Post LabDocument2 pagesChem 160.1 Exer 9 Post LabAlthea Karmylle M. BonitaNo ratings yet
- O Heterocycles From Unsaturated Carbonyls and DimethoxycarbeneDocument10 pagesO Heterocycles From Unsaturated Carbonyls and DimethoxycarbeneSaurav PaulNo ratings yet
- Periodic Tales: A Cultural History of the Elements, from Arsenic to ZincFrom EverandPeriodic Tales: A Cultural History of the Elements, from Arsenic to ZincRating: 3.5 out of 5 stars3.5/5 (137)
- Organic Chemistry for Schools: Advanced Level and Senior High SchoolFrom EverandOrganic Chemistry for Schools: Advanced Level and Senior High SchoolNo ratings yet
- Is That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeFrom EverandIs That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeRating: 5 out of 5 stars5/5 (4)
- Chemistry for Breakfast: The Amazing Science of Everyday LifeFrom EverandChemistry for Breakfast: The Amazing Science of Everyday LifeRating: 4.5 out of 5 stars4.5/5 (90)
- Formulating, Packaging, and Marketing of Natural Cosmetic ProductsFrom EverandFormulating, Packaging, and Marketing of Natural Cosmetic ProductsNo ratings yet
- Handbook of Formulating Dermal Applications: A Definitive Practical GuideFrom EverandHandbook of Formulating Dermal Applications: A Definitive Practical GuideNo ratings yet
- The Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactFrom EverandThe Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactRating: 5 out of 5 stars5/5 (5)
- Monkeys, Myths, and Molecules: Separating Fact from Fiction, and the Science of Everyday LifeFrom EverandMonkeys, Myths, and Molecules: Separating Fact from Fiction, and the Science of Everyday LifeRating: 4 out of 5 stars4/5 (1)
- The Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsFrom EverandThe Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsRating: 4 out of 5 stars4/5 (146)
- AP® Chemistry Crash Course, For the 2020 Exam, Book + Online: Get a Higher Score in Less TimeFrom EverandAP® Chemistry Crash Course, For the 2020 Exam, Book + Online: Get a Higher Score in Less TimeRating: 5 out of 5 stars5/5 (1)
- AP Chemistry Flashcards, Fourth Edition: Up-to-Date Review and PracticeFrom EverandAP Chemistry Flashcards, Fourth Edition: Up-to-Date Review and PracticeNo ratings yet
- The Regenerative Grower's Guide to Garden Amendments: Using Locally Sourced Materials to Make Mineral and Biological Extracts and FermentsFrom EverandThe Regenerative Grower's Guide to Garden Amendments: Using Locally Sourced Materials to Make Mineral and Biological Extracts and FermentsRating: 5 out of 5 stars5/5 (3)
- The Elements We Live By: How Iron Helps Us Breathe, Potassium Lets Us See, and Other Surprising Superpowers of the Periodic TableFrom EverandThe Elements We Live By: How Iron Helps Us Breathe, Potassium Lets Us See, and Other Surprising Superpowers of the Periodic TableRating: 3.5 out of 5 stars3.5/5 (22)
- Taste: Surprising Stories and Science About Why Food Tastes GoodFrom EverandTaste: Surprising Stories and Science About Why Food Tastes GoodRating: 3 out of 5 stars3/5 (20)
- The Periodic Table: A Very Short IntroductionFrom EverandThe Periodic Table: A Very Short IntroductionRating: 4.5 out of 5 stars4.5/5 (3)
- Tribology: Friction and Wear of Engineering MaterialsFrom EverandTribology: Friction and Wear of Engineering MaterialsRating: 5 out of 5 stars5/5 (1)
- Water-Based Paint Formulations, Vol. 3From EverandWater-Based Paint Formulations, Vol. 3Rating: 4.5 out of 5 stars4.5/5 (6)
- Ingredients: A Visual Exploration of 75 Additives & 25 Food ProductsFrom EverandIngredients: A Visual Exploration of 75 Additives & 25 Food ProductsRating: 4 out of 5 stars4/5 (1)
- The Periodic Table of Elements - Alkali Metals, Alkaline Earth Metals and Transition Metals | Children's Chemistry BookFrom EverandThe Periodic Table of Elements - Alkali Metals, Alkaline Earth Metals and Transition Metals | Children's Chemistry BookNo ratings yet
- Chemistry for Breakfast: The Amazing Science of Everyday LifeFrom EverandChemistry for Breakfast: The Amazing Science of Everyday LifeRating: 4.5 out of 5 stars4.5/5 (14)
- Bioplastics: A Home Inventors HandbookFrom EverandBioplastics: A Home Inventors HandbookRating: 4 out of 5 stars4/5 (2)
- High School Chemistry: Comprehensive Content for High School ChemistryFrom EverandHigh School Chemistry: Comprehensive Content for High School ChemistryNo ratings yet
- The Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactFrom EverandThe Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactRating: 5 out of 5 stars5/5 (1)
- Chemistry: a QuickStudy Laminated Reference GuideFrom EverandChemistry: a QuickStudy Laminated Reference GuideRating: 5 out of 5 stars5/5 (1)