You might also like
- Kinetics of The Autocatalytic Deposition of Ni-P Alloys in Ammoniacal SolutionsDocument8 pagesKinetics of The Autocatalytic Deposition of Ni-P Alloys in Ammoniacal SolutionsAngelo VarelaNo ratings yet
- Removal of Lead From Crude Antimony by Using Napo As Lead Elimination ReagentDocument7 pagesRemoval of Lead From Crude Antimony by Using Napo As Lead Elimination ReagentTacachiri Chocamani JaimeNo ratings yet
- Effects of Synthesis Conditions On The Structural and Electrochemical PropertiesDocument5 pagesEffects of Synthesis Conditions On The Structural and Electrochemical PropertiesjoseNo ratings yet
- Rivers WaterDocument5 pagesRivers WaterBilal JuttNo ratings yet
- 19PN08Document29 pages19PN08karthik soundar NAGARAJNo ratings yet
- 2010 - Preparation of Nanocrystalline Lithium Niobate Powders at Low TemperatureDocument6 pages2010 - Preparation of Nanocrystalline Lithium Niobate Powders at Low Temperature13408169705No ratings yet
- Materials: Hydrothermal Synthesis of Metal Oxide Nanoparticles in Supercritical WaterDocument24 pagesMaterials: Hydrothermal Synthesis of Metal Oxide Nanoparticles in Supercritical WaterSarath KumarNo ratings yet
- JNCRS Vol3 01-05Document5 pagesJNCRS Vol3 01-05Pratikshya PriyadarshiniNo ratings yet
- Evaluations of Discharge Capacity and Cycle Stability For A Graphene-AddedDocument5 pagesEvaluations of Discharge Capacity and Cycle Stability For A Graphene-AddedjoseNo ratings yet
- 1 s2.0 S0925838823006862 mmc1Document25 pages1 s2.0 S0925838823006862 mmc1J MrNo ratings yet
- Accepted Manuscript: Applied Surface ScienceDocument21 pagesAccepted Manuscript: Applied Surface ScienceAlina BurduceaNo ratings yet
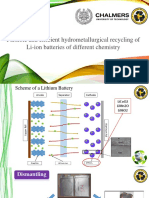
- Flexible and Efficient Hydrometallurgical Recycling of Li-Ion Batteries of Different ChemistryDocument46 pagesFlexible and Efficient Hydrometallurgical Recycling of Li-Ion Batteries of Different Chemistrytaufiq_hidayat_1982No ratings yet
- Materials Research Bulletin: Yang You, Long Wan, Shiying Zhang, Difa XuDocument5 pagesMaterials Research Bulletin: Yang You, Long Wan, Shiying Zhang, Difa XuChemist.AlchemistNo ratings yet
- TS. Trương Thái Giang - Hội thảo khoa học Đại học Thành ĐôDocument10 pagesTS. Trương Thái Giang - Hội thảo khoa học Đại học Thành ĐôLưu Thu HàNo ratings yet
- MgO Effect TGDocument3 pagesMgO Effect TGBiswanath senNo ratings yet
- Matecconf cct2016 01007Document7 pagesMatecconf cct2016 01007Abdulrahman JradiNo ratings yet
- Elaboration and Nanostructural Study of Pure and Al Doped Zno NanopowdersDocument5 pagesElaboration and Nanostructural Study of Pure and Al Doped Zno NanopowdersGabriela PlaiasuNo ratings yet
- Poll Res-61 - Published Paper - Priyanka WaditakeDocument6 pagesPoll Res-61 - Published Paper - Priyanka WaditakePrashantNo ratings yet
- JMC Co3O4forsupercapaDocument7 pagesJMC Co3O4forsupercapamac.zahara3344No ratings yet
- Selective Oxidation of Styrene To Benzaldehyde byDocument7 pagesSelective Oxidation of Styrene To Benzaldehyde bywiam wiamNo ratings yet
- Ion ExchangeDocument3 pagesIon ExchangeelftahtasakalNo ratings yet
- Tellier 2007Document14 pagesTellier 2007Venu CharyNo ratings yet
- Mirkhani2009 Article PhotocatalyticDegradationOfAzoDocument10 pagesMirkhani2009 Article PhotocatalyticDegradationOfAzoAjit Kumar DhankaNo ratings yet
- synthesis of α-MnO2 using KIO4Document3 pagessynthesis of α-MnO2 using KIO4Đoàn Trí KhoaNo ratings yet
- Hidrotalcite 2Document1 pageHidrotalcite 2Amir SetiadiNo ratings yet
- Electrocoagulation Treatment For Removal of Color and Chemical Oxygen Demand in Landfill Leachate Using Aluminum ElectrodeDocument4 pagesElectrocoagulation Treatment For Removal of Color and Chemical Oxygen Demand in Landfill Leachate Using Aluminum ElectrodeDarth FrootLoopsNo ratings yet
- Ion ExchangeDocument4 pagesIon ExchangeBhinitha ChandrasagaranNo ratings yet
- 1783 Pui PDFDocument9 pages1783 Pui PDFKhuyen VoNo ratings yet
- SEMINAR On NanocompositesDocument30 pagesSEMINAR On NanocompositesRama GaurNo ratings yet
- 1 s2.0 S0955221905009477 MainDocument7 pages1 s2.0 S0955221905009477 MainTio Putra WendariNo ratings yet
- Microestructure and Composition of Rare Earth...Document6 pagesMicroestructure and Composition of Rare Earth...Daiana NavarreteNo ratings yet
- Materials 12 00702Document16 pagesMaterials 12 00702Manish ShrungareNo ratings yet
- Comparision of The Flow in Co-Rotating and Counter-Rotating TwinscrewDocument11 pagesComparision of The Flow in Co-Rotating and Counter-Rotating TwinscrewChauNo ratings yet
- Powder Technology: Prita Pant Sarangi, S.R. Vadera, M.K. Patra, N.N. GhoshDocument6 pagesPowder Technology: Prita Pant Sarangi, S.R. Vadera, M.K. Patra, N.N. Ghoshehagar60No ratings yet
- JMC Co3O4forsupercapaDocument7 pagesJMC Co3O4forsupercapaDreen DiazNo ratings yet
- 10.1515 - Epoly 2013 0126Document8 pages10.1515 - Epoly 2013 0126poonam4927No ratings yet
- Highly Efficient Al-Doped Zno: Ag Catalyst For Rb19 Photocatalytic Degradation: Microwave-Assisted Synthesis and CharacterizationDocument9 pagesHighly Efficient Al-Doped Zno: Ag Catalyst For Rb19 Photocatalytic Degradation: Microwave-Assisted Synthesis and CharacterizationAnaGomezNo ratings yet
- Chaudhari2013 Article StructuralMagneticAndDielectriDocument6 pagesChaudhari2013 Article StructuralMagneticAndDielectritahaNo ratings yet
- Geng 2016Document5 pagesGeng 2016Septia Kurniawati ArifahNo ratings yet
- Materials 04 00153Document16 pagesMaterials 04 00153Gaël MOUKENGUENo ratings yet
- LDH SpectraDocument7 pagesLDH SpectraShamsheer KhanNo ratings yet
- 1 s2.0 S0304885315305205 MainDocument6 pages1 s2.0 S0304885315305205 MainHuckkey HuNo ratings yet
- The Effects of Temperature On The Conversion of Li2CO3 To LiOH in A CA (OH) 2 SuspensionDocument6 pagesThe Effects of Temperature On The Conversion of Li2CO3 To LiOH in A CA (OH) 2 SuspensionARREDONDO OLIVOS CARLOS ENRIQUENo ratings yet
- Structure and Catalytic Activity of La Feo System (X 0.00, 0.05, 0.10, 0.15, 0.20, 0.25, 0.35) For The No Co ReactionDocument16 pagesStructure and Catalytic Activity of La Feo System (X 0.00, 0.05, 0.10, 0.15, 0.20, 0.25, 0.35) For The No Co Reactionmompou88No ratings yet
- Identification of MICDocument12 pagesIdentification of MICShesharam ChouhanNo ratings yet
- ZN (II) Formate Bpy Coordination Polymer 2016Document5 pagesZN (II) Formate Bpy Coordination Polymer 2016Legeek GamingNo ratings yet
- (CoNi) MN-LDH Nanosheets On MWCNT For OER SIDocument18 pages(CoNi) MN-LDH Nanosheets On MWCNT For OER SICB Dong SuwonNo ratings yet
- Chemical and Physical Properties of Fluxes For SAW of Low-Carbon SteelsDocument20 pagesChemical and Physical Properties of Fluxes For SAW of Low-Carbon SteelsAdam Al AufaNo ratings yet
- Synthesis and Piezoelectric Properties of (Na0.5Bi0.5) 0.94Ba0.06TiO3Document5 pagesSynthesis and Piezoelectric Properties of (Na0.5Bi0.5) 0.94Ba0.06TiO3héma tologieNo ratings yet
- Optimization of Mechanical Behavior of Ni-P Nanocomposite Coatings Using Taguchi ApproachDocument12 pagesOptimization of Mechanical Behavior of Ni-P Nanocomposite Coatings Using Taguchi ApproachTJPRC PublicationsNo ratings yet
- Synthesis and Characterization of Tetrakis2-AminoDocument7 pagesSynthesis and Characterization of Tetrakis2-AminoGerónimo PerazzoNo ratings yet
- 1 s2.0 S0022024824000186 MainDocument22 pages1 s2.0 S0022024824000186 Mainwb9269xkzkNo ratings yet
- TS. Trương Thái Giang - Hội thảo khoa học Đại học Thành ĐôDocument9 pagesTS. Trương Thái Giang - Hội thảo khoa học Đại học Thành ĐôLưu Thu HàNo ratings yet
- Senol 2015Document15 pagesSenol 2015Rajesh KumarNo ratings yet
- Synthesis Characterization of Photoactive Complex and Study Its Photochemical ReactionDocument4 pagesSynthesis Characterization of Photoactive Complex and Study Its Photochemical ReactionOSCAR DANIEL ARREDONDO GAÑANNo ratings yet
- Singh e Rakesh, 2009Document3 pagesSingh e Rakesh, 2009Mariana RochaNo ratings yet
- Study of Reflection Loss in Ku Band by CNM Decorated With Metal Nano ParticlesDocument10 pagesStudy of Reflection Loss in Ku Band by CNM Decorated With Metal Nano ParticlesIJRASETPublicationsNo ratings yet
- 602-Article Text-5769-3-10-20150204Document8 pages602-Article Text-5769-3-10-20150204Arrei x5No ratings yet
- Iron Metabolism: From Molecular Mechanisms to Clinical ConsequencesFrom EverandIron Metabolism: From Molecular Mechanisms to Clinical ConsequencesRating: 5 out of 5 stars5/5 (1)
- Schrodinger Wave EquationsDocument6 pagesSchrodinger Wave EquationsksksvtNo ratings yet
- Definition of Logistics ManagementDocument4 pagesDefinition of Logistics ManagementzamaneNo ratings yet
- Grocery GatewayDocument2 pagesGrocery GatewayKumari Mohan0% (2)
- Eurolite Led TMH 7 PDFDocument2 pagesEurolite Led TMH 7 PDFSarahNo ratings yet
- Lesson 24 - Laminate Modeling - Rev C PDFDocument20 pagesLesson 24 - Laminate Modeling - Rev C PDFraduga_fbNo ratings yet
- Hot Process Liquid SoapmakingDocument11 pagesHot Process Liquid SoapmakingPanacea PharmaNo ratings yet
- SC4860-48120 (-MPV) User Manual EN 202107Document23 pagesSC4860-48120 (-MPV) User Manual EN 202107Nattachai BoonchooNo ratings yet
- Load ScheduleDocument8 pagesLoad SchedulemerebookNo ratings yet
- Methanol Technologies of Tkis: A Brief OverviewDocument12 pagesMethanol Technologies of Tkis: A Brief OverviewMuhammad NaeemNo ratings yet
- Stahl - PUDSDocument29 pagesStahl - PUDSsusanasusana_No ratings yet
- How To Build A GreenhouseDocument67 pagesHow To Build A GreenhouseBolarinwaNo ratings yet
- NSTM.512v0.FANS.S9086 RS STM 010 CH 512R21Sep99Document60 pagesNSTM.512v0.FANS.S9086 RS STM 010 CH 512R21Sep99jwfqcw74mfNo ratings yet
- Annual Syllabus Class 7 1Document3 pagesAnnual Syllabus Class 7 1Ni shNo ratings yet
- Wind Load CompututationsDocument31 pagesWind Load Compututationskim suarezNo ratings yet
- Money, Interest, and Inflation: Answers To CheckpointsDocument16 pagesMoney, Interest, and Inflation: Answers To Checkpointsb96309No ratings yet
- Iron FistDocument2 pagesIron FistVictor PileggiNo ratings yet
- Chem 3052 CHAPTER 7 (Nuclear Magnetic Resonance Spectroscopy (NMR) )Document6 pagesChem 3052 CHAPTER 7 (Nuclear Magnetic Resonance Spectroscopy (NMR) )ashenafiNo ratings yet
- Electro-Mechanical SectorDocument22 pagesElectro-Mechanical SectorKen LeeNo ratings yet
- DRS Rev.0 GTP-TR1!01!002 Condensate RecyclingDocument4 pagesDRS Rev.0 GTP-TR1!01!002 Condensate RecyclingBalasubramanianNo ratings yet
- Chemical Resistance TableDocument6 pagesChemical Resistance TableEdgarDavidDiazCamposNo ratings yet
- Augustine's Confessions - Philosophy in AutobiographyDocument241 pagesAugustine's Confessions - Philosophy in AutobiographyAlfonso Flórez100% (2)
- Samuelson and Nordhaus ch22 PDFDocument30 pagesSamuelson and Nordhaus ch22 PDFVictor ManatadNo ratings yet
- Circle Theorems, Class 10Document7 pagesCircle Theorems, Class 10Susan MathewNo ratings yet
- Oertel - Extracts From The Jāiminīya-Brāhma A and Upanishad-Brāhma A, Parallel To Passages of TheDocument20 pagesOertel - Extracts From The Jāiminīya-Brāhma A and Upanishad-Brāhma A, Parallel To Passages of Thespongebob2812No ratings yet
- 6RA80 Quick Commissioning Without TachoDocument7 pages6RA80 Quick Commissioning Without TachoBaldev SinghNo ratings yet
- EE 411-Digital Signal Processing-Muhammad TahirDocument3 pagesEE 411-Digital Signal Processing-Muhammad TahirQasim FarooqNo ratings yet
- ECE 374 - Part - 1c - S2017Document37 pagesECE 374 - Part - 1c - S2017Zakaria ElwalilyNo ratings yet
- Shadow UAV HandbookDocument57 pagesShadow UAV HandbookGasMaskBob100% (2)
- Procrustes AlgorithmDocument11 pagesProcrustes AlgorithmShoukkathAliNo ratings yet
- Homoeopathy and MigraineDocument4 pagesHomoeopathy and MigraineEditor IJTSRDNo ratings yet