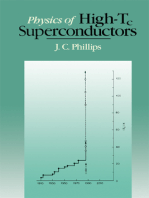
You might also like
- 2016 JMCA AceramicpolymercompositesolidelectrolyteforDocument6 pages2016 JMCA AceramicpolymercompositesolidelectrolyteforSteven KmiecNo ratings yet
- 2012 Jakubas Bis (Imidazolium) Pentachloroantimonate (III)Document4 pages2012 Jakubas Bis (Imidazolium) Pentachloroantimonate (III)Nacho Delgado FerreiroNo ratings yet
- PCCP BDT ThermoDocument7 pagesPCCP BDT ThermoJULIO CESAR XAVIER CONCEICAO MARRANo ratings yet
- DFT Study On Elastic and Pirzoelectric Properties of Tetragonal BaTiO3Document6 pagesDFT Study On Elastic and Pirzoelectric Properties of Tetragonal BaTiO3Ricardo MagallanesNo ratings yet
- Journal of Alloys and CompoundsDocument12 pagesJournal of Alloys and CompoundsTalita SouzaNo ratings yet
- Perovskite BagusDocument57 pagesPerovskite BagusaditngrhNo ratings yet
- Dalton Transactions: PaperDocument11 pagesDalton Transactions: PaperGustavo GuillénNo ratings yet
- Zhou 2018 Mater. Res. Express 5 095510Document11 pagesZhou 2018 Mater. Res. Express 5 095510Víctor SaldarriagaNo ratings yet
- Physics Letters A: Manish Kumar, S. Shankar, Brijmohan, Shiv Kumar, O.P. Thakur, Anup K. GhoshDocument8 pagesPhysics Letters A: Manish Kumar, S. Shankar, Brijmohan, Shiv Kumar, O.P. Thakur, Anup K. GhoshMAPACHE 1260No ratings yet
- Materials Research BulletinDocument7 pagesMaterials Research BulletinSamah SamahNo ratings yet
- Atypical Multiferroicity of HoCrO3 in Bulk and Film - Ghosh - Journal of Materials Chemistry CDocument6 pagesAtypical Multiferroicity of HoCrO3 in Bulk and Film - Ghosh - Journal of Materials Chemistry CNeeraj PanwarNo ratings yet
- Acta Materialia: Xunxiang Hu, Chad M. Parish, Kun Wang, Takaaki Koyanagi, Benjamin P. Eftink, Yutai KatohDocument11 pagesActa Materialia: Xunxiang Hu, Chad M. Parish, Kun Wang, Takaaki Koyanagi, Benjamin P. Eftink, Yutai KatohWalid BadrNo ratings yet
- Journal of The European Ceramic Society: Vincenzo Buscaglia, Clive A. Randall TDocument15 pagesJournal of The European Ceramic Society: Vincenzo Buscaglia, Clive A. Randall TAryan Singh LatherNo ratings yet
- Paper: Systematic Analysis of Electron Energy-Loss Near-Edge Structures in Li-Ion Battery MaterialsDocument10 pagesPaper: Systematic Analysis of Electron Energy-Loss Near-Edge Structures in Li-Ion Battery MaterialsSorina CretuNo ratings yet
- ST-BT Nanocomposite - JMatChem C 2017Document9 pagesST-BT Nanocomposite - JMatChem C 2017Cristina CiomagaNo ratings yet
- Enhanced Piezoelectricity and Nature of Electric-Field Induced Structural Phase Transformation in Textured Lead-Free Piezoelectric Na0.5Bi0.5Tio3-Batio3 CeramicsDocument6 pagesEnhanced Piezoelectricity and Nature of Electric-Field Induced Structural Phase Transformation in Textured Lead-Free Piezoelectric Na0.5Bi0.5Tio3-Batio3 CeramicsSamah SamahNo ratings yet
- Crystals 13 00255 v2Document13 pagesCrystals 13 00255 v2Menjamin SalasNo ratings yet
- Paper Dalton Transactions 2014Document16 pagesPaper Dalton Transactions 2014vkphyNo ratings yet
- 10 1039@C9TC01456JDocument7 pages10 1039@C9TC01456JElnurNo ratings yet
- Ferroelectric NotesDocument6 pagesFerroelectric Noteschvar80100% (1)
- Beyond The Molecular Orbital Conception of Electronically Excited States Through The Quantum Theory of Atoms in MoleculesDocument11 pagesBeyond The Molecular Orbital Conception of Electronically Excited States Through The Quantum Theory of Atoms in MoleculesShahid Husen SakhiwalaNo ratings yet
- Theory of Antiferroelectric CrystalsDocument4 pagesTheory of Antiferroelectric CrystalsJJ SerraltaNo ratings yet
- Current Applied Physics: J. Paul Praveen, Kranti Kumar, A.R. James, T. Karthik, Saket Asthana, Dibakar DasDocument7 pagesCurrent Applied Physics: J. Paul Praveen, Kranti Kumar, A.R. James, T. Karthik, Saket Asthana, Dibakar DasSamah SamahNo ratings yet
- Barium Titanate As A Ferroelectric and Piezoelectric CeramicsDocument4 pagesBarium Titanate As A Ferroelectric and Piezoelectric CeramicsShabir KhanNo ratings yet
- Philosophical Magazine A: To Cite This Article: A. Planes, J. L. Macqueron, R. Rapacioli & G. Guénin (1990) MartensiticDocument12 pagesPhilosophical Magazine A: To Cite This Article: A. Planes, J. L. Macqueron, R. Rapacioli & G. Guénin (1990) MartensiticMuhammad Falqi YusufNo ratings yet
- Resolving Structural Contributions To The Electric-Field-Induced Strain in Lead-Free (1 X) Ba (ZR Ti) O X (Ba Ca) Tio PiezoceramicsDocument9 pagesResolving Structural Contributions To The Electric-Field-Induced Strain in Lead-Free (1 X) Ba (ZR Ti) O X (Ba Ca) Tio PiezoceramicsSamah SamahNo ratings yet
- Energies 16 01294 v2Document21 pagesEnergies 16 01294 v2EstefanyNo ratings yet
- Long-Range and Short-Range Structure of Proton-Conducting Y:BazroDocument9 pagesLong-Range and Short-Range Structure of Proton-Conducting Y:BazroAlex SpradaNo ratings yet
- Graphene 2Document5 pagesGraphene 2Science BrainsNo ratings yet
- Tutorial 8 Solution - Solid State Physics PDFDocument12 pagesTutorial 8 Solution - Solid State Physics PDFRaHuL MuSaLe100% (1)
- Theory of A Quantum Critical Phenomenon in A Topological Insulator: (3+1) Dimensional Quantum Electrodynamics in SolidsDocument6 pagesTheory of A Quantum Critical Phenomenon in A Topological Insulator: (3+1) Dimensional Quantum Electrodynamics in SolidsNetto HolandaNo ratings yet
- 2013 Li Pyroelectric and Electrocaloric MaterialsDocument15 pages2013 Li Pyroelectric and Electrocaloric MaterialsNacho Delgado FerreiroNo ratings yet
- 10.1515 - Zna 2016 0149Document10 pages10.1515 - Zna 2016 0149rajanadarajanNo ratings yet
- On The Diameter Dependence of Metal-Nanowire Schottky Barrier HeightDocument8 pagesOn The Diameter Dependence of Metal-Nanowire Schottky Barrier HeightSeamNo ratings yet
- 00150193.2019.1592495 BNT BT BY SOLID STATEDocument9 pages00150193.2019.1592495 BNT BT BY SOLID STATEAditi SharmaNo ratings yet
- Bio Codoped BCZTDocument10 pagesBio Codoped BCZTRachna SelvamaniNo ratings yet
- CHPT 06 Piezoelectric MaterialsDocument29 pagesCHPT 06 Piezoelectric Materialsjandtmiller8057100% (1)
- 1 s2.0 S135964542200091X MainDocument8 pages1 s2.0 S135964542200091X Mainzj xiaoNo ratings yet
- Theoretical Investigations of The Bulk Modulus in The Tetra-Cubic Transition of Pbtio Material Renan A. P. Ribeiro and Sergio R. de LázaroDocument6 pagesTheoretical Investigations of The Bulk Modulus in The Tetra-Cubic Transition of Pbtio Material Renan A. P. Ribeiro and Sergio R. de LázaroYangWenNo ratings yet
- RSC Advances: PaperDocument7 pagesRSC Advances: PaperPedro Elias Romero NietoNo ratings yet
- Acta Materialia: Full Length ArticleDocument15 pagesActa Materialia: Full Length Articleshouxun JiNo ratings yet
- Journal of Alloys and Compounds: Wei Li, Zhijun Xu, Ruiqing Chu, Peng Fu, Guozhong ZangDocument4 pagesJournal of Alloys and Compounds: Wei Li, Zhijun Xu, Ruiqing Chu, Peng Fu, Guozhong ZangSamah SamahNo ratings yet
- Stripesin YBCODocument2 pagesStripesin YBCOIon IvanNo ratings yet
- Lecture 12Document33 pagesLecture 12Daniele PasseroneNo ratings yet
- Articulo NanotecnlogiaDocument18 pagesArticulo NanotecnlogiaHector ZaNo ratings yet
- Neutron and NeutronDocument10 pagesNeutron and Neutronsamuel_07No ratings yet
- Berrada 2023 Laser Phys. Lett. 20 085201Document7 pagesBerrada 2023 Laser Phys. Lett. 20 085201Hichem El EuchNo ratings yet
- Broadband Dielectric Response of CaCu3Ti4O12Document8 pagesBroadband Dielectric Response of CaCu3Ti4O12Alexandr BabaskinNo ratings yet
- Journal of The American Ceramic Society - 2017 - Feng - Relaxor Nature in Ba5RZr3Nb7O30 R La ND SM TetragonalDocument9 pagesJournal of The American Ceramic Society - 2017 - Feng - Relaxor Nature in Ba5RZr3Nb7O30 R La ND SM Tetragonalton nu thanh phuongNo ratings yet
- Synthesis and Characterizations of BNT-BT-KNN Ceramics For Energy Storage ApplicationsDocument15 pagesSynthesis and Characterizations of BNT-BT-KNN Ceramics For Energy Storage ApplicationsM CHANDRASEKHARNo ratings yet
- Self-Consistent Phonon Calculations of Lattice DynDocument12 pagesSelf-Consistent Phonon Calculations of Lattice DynsujatharajanNo ratings yet
- Dilectric Constant IMPDocument12 pagesDilectric Constant IMPPratikshya PriyadarshiniNo ratings yet
- C0JM04058D 4861..4868Document8 pagesC0JM04058D 4861..4868Aryan Singh LatherNo ratings yet
- BZT Tun - Ferroelectrics 2009Document9 pagesBZT Tun - Ferroelectrics 2009Cristina CiomagaNo ratings yet
- ARTIGO 7 ESTRUTURA DOS NANOTUBOS Peralta-Inga2003Document13 pagesARTIGO 7 ESTRUTURA DOS NANOTUBOS Peralta-Inga2003ElesseaNo ratings yet
- Three Center and 4 Electron BondDocument12 pagesThree Center and 4 Electron BondaziahmughalNo ratings yet
- Theoretical Studies of The ParamagneticDocument8 pagesTheoretical Studies of The Paramagneticimti8509No ratings yet
- 2 - Transport Properties of The Pseudo-BinaryDocument4 pages2 - Transport Properties of The Pseudo-BinarySena KulaksızNo ratings yet
- Journal of Materials Chemistry C: Feature ArticleDocument21 pagesJournal of Materials Chemistry C: Feature ArticleMoamen MohamedNo ratings yet
- Silo Dryers: Mepu - Farmer S First Choice Mepu To Suit Every UserDocument2 pagesSilo Dryers: Mepu - Farmer S First Choice Mepu To Suit Every UserTahir Güçlü100% (1)
- Using A Wet Film ThicknessDocument2 pagesUsing A Wet Film ThicknesssanoopvkNo ratings yet
- Environmental and Sustainability Issues - 1Document21 pagesEnvironmental and Sustainability Issues - 121. PLT PAGALILAUAN, EDITHA MNo ratings yet
- Big Fat Lies - How The Diet Industry Is Making You Sick, Fat & PoorDocument212 pagesBig Fat Lies - How The Diet Industry Is Making You Sick, Fat & PoorangelobuffaloNo ratings yet
- Hot Process Liquid SoapmakingDocument11 pagesHot Process Liquid SoapmakingPanacea PharmaNo ratings yet
- 2.PsychoCrash Social Psy-1Document62 pages2.PsychoCrash Social Psy-1Gopika Sureshnv0% (1)
- Advanced Logic Synthesis: Multiple Choice QuestionsDocument16 pagesAdvanced Logic Synthesis: Multiple Choice QuestionsmanojkumarNo ratings yet
- Q3 Module 15Document33 pagesQ3 Module 15jovielyn kathley manaloNo ratings yet
- Mold Maintenance StepDocument0 pagesMold Maintenance StepMonica JoynerNo ratings yet
- NID DATPrelimsTestPaper2018 BDesDocument24 pagesNID DATPrelimsTestPaper2018 BDesManaswini ReddyNo ratings yet
- Ruger MKIIDocument1 pageRuger MKIIMike Pape100% (1)
- Academic Program Required Recommended Academic Program Required RecommendedDocument1 pageAcademic Program Required Recommended Academic Program Required Recommendedonur scribdNo ratings yet
- Black Mamba Vs Mongoose Vs King Cobra Vs Komodo Vs PhythonDocument44 pagesBlack Mamba Vs Mongoose Vs King Cobra Vs Komodo Vs PhythonmarcNo ratings yet
- Sample Menu FinalDocument1 pageSample Menu FinalChris HughesNo ratings yet
- DysphagiaDocument4 pagesDysphagiaMicaNo ratings yet
- Sdo Tle Grade 8 Dressmaking 2nd Q Week 1 8 1Document64 pagesSdo Tle Grade 8 Dressmaking 2nd Q Week 1 8 1Maggie De jesusNo ratings yet
- T60n02rg PDFDocument8 pagesT60n02rg PDFsandor9116100% (2)
- Acc05 SCG116Document42 pagesAcc05 SCG116Hilal HazaaNo ratings yet
- 2006 - Dong Et Al - Bulk and Dispersed Aqueous Phase Behavior of PhytantriolDocument7 pages2006 - Dong Et Al - Bulk and Dispersed Aqueous Phase Behavior of PhytantriolHe ZeeNo ratings yet
- Quran On GeologyDocument10 pagesQuran On GeologyMM NabeelNo ratings yet
- Acute Coronary Syndrome: Diagnosis and Initial Management: Each YearDocument9 pagesAcute Coronary Syndrome: Diagnosis and Initial Management: Each YearGabriela Pacheco0% (1)
- VENUS e CatalogueDocument38 pagesVENUS e CatalogueGanesh BabuNo ratings yet
- Pocket Book AGDocument67 pagesPocket Book AGsudiraharjaNo ratings yet
- HYW-17 T5 S5: Industrial Range MobileDocument6 pagesHYW-17 T5 S5: Industrial Range MobileghostshotNo ratings yet
- Aipl2009 V120000001Document7 pagesAipl2009 V120000001Olof HedinNo ratings yet
- Grocery GatewayDocument2 pagesGrocery GatewayKumari Mohan0% (2)
- Lesson 24 - Laminate Modeling - Rev C PDFDocument20 pagesLesson 24 - Laminate Modeling - Rev C PDFraduga_fbNo ratings yet
- Itinerary - State 2010Document3 pagesItinerary - State 2010purest123No ratings yet
- Module III Rural MarketingDocument30 pagesModule III Rural MarketingNikita YadavNo ratings yet
- X Glo LED Strip Lighting For Tunnelling BrochureDocument6 pagesX Glo LED Strip Lighting For Tunnelling BrochureJOSE HUAMANINo ratings yet
- Dark Matter and the Dinosaurs: The Astounding Interconnectedness of the UniverseFrom EverandDark Matter and the Dinosaurs: The Astounding Interconnectedness of the UniverseRating: 3.5 out of 5 stars3.5/5 (69)
- The Rise and Fall of the Dinosaurs: A New History of a Lost WorldFrom EverandThe Rise and Fall of the Dinosaurs: A New History of a Lost WorldRating: 4 out of 5 stars4/5 (597)
- Alex & Me: How a Scientist and a Parrot Discovered a Hidden World of Animal Intelligence—and Formed a Deep Bond in the ProcessFrom EverandAlex & Me: How a Scientist and a Parrot Discovered a Hidden World of Animal Intelligence—and Formed a Deep Bond in the ProcessNo ratings yet
- The Other End of the Leash: Why We Do What We Do Around DogsFrom EverandThe Other End of the Leash: Why We Do What We Do Around DogsRating: 5 out of 5 stars5/5 (65)
- The Ancestor's Tale: A Pilgrimage to the Dawn of EvolutionFrom EverandThe Ancestor's Tale: A Pilgrimage to the Dawn of EvolutionRating: 4 out of 5 stars4/5 (812)
- The Lives of Bees: The Untold Story of the Honey Bee in the WildFrom EverandThe Lives of Bees: The Untold Story of the Honey Bee in the WildRating: 4.5 out of 5 stars4.5/5 (44)
- Fire Season: Field Notes from a Wilderness LookoutFrom EverandFire Season: Field Notes from a Wilderness LookoutRating: 4 out of 5 stars4/5 (142)
- The Hawk's Way: Encounters with Fierce BeautyFrom EverandThe Hawk's Way: Encounters with Fierce BeautyRating: 4.5 out of 5 stars4.5/5 (19)
- The Revolutionary Genius of Plants: A New Understanding of Plant Intelligence and BehaviorFrom EverandThe Revolutionary Genius of Plants: A New Understanding of Plant Intelligence and BehaviorRating: 4.5 out of 5 stars4.5/5 (137)
- When You Find Out the World Is Against You: And Other Funny Memories About Awful MomentsFrom EverandWhen You Find Out the World Is Against You: And Other Funny Memories About Awful MomentsRating: 3.5 out of 5 stars3.5/5 (13)
- Spoiled Rotten America: Outrages of Everyday LifeFrom EverandSpoiled Rotten America: Outrages of Everyday LifeRating: 3 out of 5 stars3/5 (19)
- Roxane Gay & Everand Originals: My Year of Psychedelics: Lessons on Better LivingFrom EverandRoxane Gay & Everand Originals: My Year of Psychedelics: Lessons on Better LivingRating: 5 out of 5 stars5/5 (5)
- Roxane Gay & Everand Originals: My Year of Psychedelics: Lessons on Better LivingFrom EverandRoxane Gay & Everand Originals: My Year of Psychedelics: Lessons on Better LivingRating: 3.5 out of 5 stars3.5/5 (35)
- Come Back, Como: Winning the Heart of a Reluctant DogFrom EverandCome Back, Como: Winning the Heart of a Reluctant DogRating: 3.5 out of 5 stars3.5/5 (10)
- Darwin's Doubt: The Explosive Origin of Animal Life and the Case for Intelligent DesignFrom EverandDarwin's Doubt: The Explosive Origin of Animal Life and the Case for Intelligent DesignRating: 4 out of 5 stars4/5 (19)
- The Hidden Life of Trees: What They Feel, How They CommunicateFrom EverandThe Hidden Life of Trees: What They Feel, How They CommunicateRating: 4 out of 5 stars4/5 (1003)
- World of Wonders: In Praise of Fireflies, Whale Sharks, and Other AstonishmentsFrom EverandWorld of Wonders: In Praise of Fireflies, Whale Sharks, and Other AstonishmentsRating: 4 out of 5 stars4/5 (223)
- Wayfinding: The Science and Mystery of How Humans Navigate the WorldFrom EverandWayfinding: The Science and Mystery of How Humans Navigate the WorldRating: 4.5 out of 5 stars4.5/5 (18)