You might also like
- Lab Report 05Document20 pagesLab Report 05DewNo ratings yet
- COMPASS: A Tool For Comparison of Multiple Protein Alignments With Assessment of Statistical SignificanceDocument20 pagesCOMPASS: A Tool For Comparison of Multiple Protein Alignments With Assessment of Statistical SignificanceВалерий БжезинскийNo ratings yet
- PSI-BLAST Tutorial - Comparative Genomics-For Term PaperDocument9 pagesPSI-BLAST Tutorial - Comparative Genomics-For Term PaperDevinder KaurNo ratings yet
- PSIPREDDocument8 pagesPSIPREDVaibhavi AwaleNo ratings yet
- Reliability 7Document10 pagesReliability 7Shampa SenNo ratings yet
- BLAST Glossary With HighlightsDocument9 pagesBLAST Glossary With Highlightsimran47No ratings yet
- Multiple Sequence Alignment MSADocument8 pagesMultiple Sequence Alignment MSAAnonymous nUiP53SNo ratings yet
- Delta Blast PDFDocument14 pagesDelta Blast PDFHark SranNo ratings yet
- Theory: Sequence Alignment Is A Process of Aligning Two Sequences To Achieve Maximum Levels ofDocument5 pagesTheory: Sequence Alignment Is A Process of Aligning Two Sequences To Achieve Maximum Levels ofGopi ShankarNo ratings yet
- Multiple Sequence AlignmentsDocument9 pagesMultiple Sequence Alignmentsvanigo1824No ratings yet
- Pattern Recognition 1Document5 pagesPattern Recognition 1Tejinder SinghNo ratings yet
- Unit2 2Document30 pagesUnit2 2sadhanaaNo ratings yet
- Lab Report 3 BioinformaticsDocument18 pagesLab Report 3 BioinformaticsRabiatul Adawiyah HasbullahNo ratings yet
- NIH Public Access: Author ManuscriptDocument19 pagesNIH Public Access: Author Manuscriptantonio.rosato1No ratings yet
- 2010 Proteins 78 118-134Document17 pages2010 Proteins 78 118-134mbrylinskiNo ratings yet
- Metamotifs - A Generative Model For Building Families of Nucleotide Position Weight MatricesDocument16 pagesMetamotifs - A Generative Model For Building Families of Nucleotide Position Weight Matricesanon_174376942No ratings yet
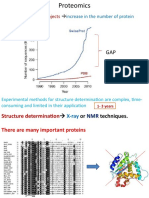
- Genome Sequencing Projects: Increase in The Number of Protein SequencesDocument27 pagesGenome Sequencing Projects: Increase in The Number of Protein SequencesBiju ThomasNo ratings yet
- H ' I P P A: MM S Nterpolation of Rotiens For Rofile NalysisDocument10 pagesH ' I P P A: MM S Nterpolation of Rotiens For Rofile NalysisijcseitNo ratings yet
- Lab Manual 6th Sem - NewDocument53 pagesLab Manual 6th Sem - NewAnbarasan SivarajNo ratings yet
- Blast ClustalDocument87 pagesBlast ClustalalexdexterNo ratings yet
- Blast 2 S, A New Tool For Comparing Protein and Nucleotide SequencesDocument4 pagesBlast 2 S, A New Tool For Comparing Protein and Nucleotide SequencesXâlmañ KháñNo ratings yet
- Merin 1Document10 pagesMerin 1merineb002No ratings yet
- Praline 3Document18 pagesPraline 3CRISTIAN GABRIEL ZAMBRANO VEGANo ratings yet
- Methods For Applying Multiple Sequence AlignmentDocument17 pagesMethods For Applying Multiple Sequence AlignmentacezeeshanNo ratings yet
- Efficient Protein Interaction Hotspot Identification in ProteinsDocument4 pagesEfficient Protein Interaction Hotspot Identification in ProteinsIJERDNo ratings yet
- Lecture 3 and 4 LSM2241Document6 pagesLecture 3 and 4 LSM2241Koh Yen SinNo ratings yet
- Effects of Gap Open and Gap Extension PenaltiesDocument5 pagesEffects of Gap Open and Gap Extension PenaltiesSantosa PradanaNo ratings yet
- Variants of Blast: By-Darshana D Ghadi Roll No. - 03Document17 pagesVariants of Blast: By-Darshana D Ghadi Roll No. - 03darshana_707No ratings yet
- Unit IiiDocument27 pagesUnit IiiDr. R. K. Selvakesavan PSGRKCWNo ratings yet
- Pairwise Sequence AlignmentDocument12 pagesPairwise Sequence Alignmentvanigo1824No ratings yet
- UntitledDocument13 pagesUntitledDewNo ratings yet
- ArticleDocument11 pagesArticleSahil8No ratings yet
- FALLSEM2022-23 BIT3001 ETH VL2022230101828 Reference Material III 13-09-2022 Gap PenaltyDocument2 pagesFALLSEM2022-23 BIT3001 ETH VL2022230101828 Reference Material III 13-09-2022 Gap PenaltyTriparna PoddarNo ratings yet
- FALLSEM2022-23 BIT3001 ETH VL2022230101828 Reference Material III 13-09-2022 Gap PenaltyDocument2 pagesFALLSEM2022-23 BIT3001 ETH VL2022230101828 Reference Material III 13-09-2022 Gap PenaltyTriparna PoddarNo ratings yet
- Homolgy ModelingDocument19 pagesHomolgy ModelingJainendra JainNo ratings yet
- M.engg Strutural Bioinformatics Assignment Submitted by AnilaDocument4 pagesM.engg Strutural Bioinformatics Assignment Submitted by AnilaAnila MumtazNo ratings yet
- Homology Modeling, Also Known As Comparative Modeling ofDocument19 pagesHomology Modeling, Also Known As Comparative Modeling ofManoharNo ratings yet
- Bioinformatics in PAM AND BLOSUMDocument17 pagesBioinformatics in PAM AND BLOSUMgladson100% (15)
- 2015 Article 14 Twilight ZoneDocument11 pages2015 Article 14 Twilight Zoneadiav PakNo ratings yet
- A Study On Protein Sequence Clustering With OPTICSDocument6 pagesA Study On Protein Sequence Clustering With OPTICSJournal of ComputingNo ratings yet
- Multiple Sequence Alignment: Hamid Hamzeiy Izmir Institute of TechnologyDocument6 pagesMultiple Sequence Alignment: Hamid Hamzeiy Izmir Institute of TechnologyHamid HamzeiyNo ratings yet
- Need & Emergence of The Field: Speaker Shashi Shekhar Head of Computational Section Biowits Life SciencesDocument59 pagesNeed & Emergence of The Field: Speaker Shashi Shekhar Head of Computational Section Biowits Life SciencesIma kuro-bikeNo ratings yet
- FASTADocument4 pagesFASTADhakshayani GNo ratings yet
- 322 Refrensi 19Document4 pages322 Refrensi 19bisniskuyNo ratings yet
- Protein Remote Homology Detection-Methods and Evaluation MetricsDocument7 pagesProtein Remote Homology Detection-Methods and Evaluation MetricsInternational Journal of Innovative Science and Research TechnologyNo ratings yet
- Fasta Sequence DatabaseDocument17 pagesFasta Sequence DatabaseKlevis XhyraNo ratings yet
- Lec06 MPH46 5103Document21 pagesLec06 MPH46 5103Sabith Hasan Khan JoyNo ratings yet
- Protein SequenceDocument36 pagesProtein SequenceHarshitNo ratings yet
- 2013 J Theor Biol 328 77-88Document12 pages2013 J Theor Biol 328 77-88mbrylinskiNo ratings yet
- SESNetDocument19 pagesSESNetThales WillianNo ratings yet
- Blast 2 Sequences, A New Tool For Comparing Protein and Nucleotide SequencesDocument17 pagesBlast 2 Sequences, A New Tool For Comparing Protein and Nucleotide SequencesXâlmañ KháñNo ratings yet
- Zhou 105Document11 pagesZhou 105suryasanNo ratings yet
- Dchip, MAS e RMADocument8 pagesDchip, MAS e RMAYuri Nagamine UrataNo ratings yet
- BioinformationDocument4 pagesBioinformationRachit NarangNo ratings yet
- 18bth028 Journal BioinformaticsDocument13 pages18bth028 Journal BioinformaticsLakshya PhoolwaniNo ratings yet
- A Genetic Algorithm With Clustering For Finding Regulatory Motifs in DNA SequencesDocument5 pagesA Genetic Algorithm With Clustering For Finding Regulatory Motifs in DNA SequencesSoumyabrataBhattacharyya1994No ratings yet
- Bioinformatics Notes - 17Bt54: Module - 4Document48 pagesBioinformatics Notes - 17Bt54: Module - 4Samuel PrinceNo ratings yet
- BlastDocument18 pagesBlastJhilik PathakNo ratings yet
- Pieper NucleicAcidsRes 2004Document6 pagesPieper NucleicAcidsRes 2004ctak2022No ratings yet
- Analysis Biomolecule Lab ReportDocument7 pagesAnalysis Biomolecule Lab ReportRani anak matNo ratings yet
- Ilovepdf MergedDocument10 pagesIlovepdf MergedRani anak matNo ratings yet
- Bmy3201 Exp 4&5Document31 pagesBmy3201 Exp 4&5Rani anak matNo ratings yet
- Biomol 3Document8 pagesBiomol 3Rani anak matNo ratings yet
- Lab Report 6,7,8,9,10,11Document26 pagesLab Report 6,7,8,9,10,11Rani anak matNo ratings yet
- BIO 151C - Cell and MolecularDocument5 pagesBIO 151C - Cell and Molecularultrauncool100% (1)
- M2-Post Task: Garcia, Mikka L. (DENT1C) 11/14/22Document2 pagesM2-Post Task: Garcia, Mikka L. (DENT1C) 11/14/22mimiNo ratings yet
- Diadumene Expansion ClonalityDocument18 pagesDiadumene Expansion ClonalitylucasmracingNo ratings yet
- DLP Dna and Protein SynthesisDocument7 pagesDLP Dna and Protein SynthesisJoeric CarinanNo ratings yet
- Phage DisplayDocument26 pagesPhage DisplaySurajit BhattacharjeeNo ratings yet
- What Is Klinefelter Syndrome ?: Genetic DiseasesDocument5 pagesWhat Is Klinefelter Syndrome ?: Genetic DiseasesTang Tiong Min 郑中铭No ratings yet
- Complete List of All Branches of Biology For SSC & Banking Exams-GK Notes in PDF!Document15 pagesComplete List of All Branches of Biology For SSC & Banking Exams-GK Notes in PDF!DevendraKumarNo ratings yet
- The Principle of PyrosequencingDocument2 pagesThe Principle of PyrosequencingClau¿ R.No ratings yet
- Bio Modul U6 2010-3Document66 pagesBio Modul U6 2010-3Thuran Nathan100% (1)
- SET 2 HSC Notes Farming For The 21st CenturyDocument14 pagesSET 2 HSC Notes Farming For The 21st CenturyAlexCuiNo ratings yet
- Huntington DiseaseDocument10 pagesHuntington Diseaseapi-498055661No ratings yet
- The Biology of CancerDocument3 pagesThe Biology of CancerPianista10% (1)
- Modern Synthesis TheoryDocument6 pagesModern Synthesis Theoryxyvel balinasNo ratings yet
- Adobe Scan 19 Feb 2023Document24 pagesAdobe Scan 19 Feb 2023nagulavanchaharishNo ratings yet
- Manuscript - Editing The Genome of CropsDocument20 pagesManuscript - Editing The Genome of CropsNiharika DasNo ratings yet
- Time-Light - Hubbard - BryanDocument24 pagesTime-Light - Hubbard - BryanYisney0% (2)
- Replication 2 PostDocument51 pagesReplication 2 PostAndy TranNo ratings yet
- Phet Natural Selection - JasonDocument6 pagesPhet Natural Selection - Jasonapi-318719438No ratings yet
- DNA StructureDocument16 pagesDNA StructureMohammedKamelNo ratings yet
- Multiple Choice Questions (MCQ) Topic Quiz Cell Division, Cell Diversity and Cellular OrganisationDocument20 pagesMultiple Choice Questions (MCQ) Topic Quiz Cell Division, Cell Diversity and Cellular OrganisationyunnNo ratings yet
- General Zoology (Notes)Document6 pagesGeneral Zoology (Notes)Faith Hannah OroNo ratings yet
- The Role of Cigarette Smoke-Induced Pulmonary Vascular Endothelial Cell Apoptosis in COPDDocument15 pagesThe Role of Cigarette Smoke-Induced Pulmonary Vascular Endothelial Cell Apoptosis in COPDAbeer SallamNo ratings yet
- Study Guide Unit 2.1 - Dna, Rna, & ProteinsDocument31 pagesStudy Guide Unit 2.1 - Dna, Rna, & ProteinsGraceNo ratings yet
- 7b. Concept of RepeatabilityDocument7 pages7b. Concept of RepeatabilityNishant ShahNo ratings yet
- Molecular Biology Topics For Research PaperDocument6 pagesMolecular Biology Topics For Research Paperfzjzn694100% (1)
- Diphenylamine TestDocument16 pagesDiphenylamine Testkenneth VonNo ratings yet
- Leopard Gecko GeneticsDocument1 pageLeopard Gecko GeneticsAgustín UrangaNo ratings yet
- Evaluation of RANK/RANKL/ OPG Gene Polymorphisms in Aggressive PeriodontitisDocument8 pagesEvaluation of RANK/RANKL/ OPG Gene Polymorphisms in Aggressive PeriodontitisLU LAM THIENNo ratings yet
- ch11 Lecture (Observable Patterns of Inheritance)Document67 pagesch11 Lecture (Observable Patterns of Inheritance)kylevNo ratings yet
- Central Dogma: Prepared By: Dyan B. JumamoyDocument89 pagesCentral Dogma: Prepared By: Dyan B. JumamoyStephanna Steff Lao-un AdimNo ratings yet