You might also like
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5814)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1092)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (845)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (897)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (540)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (348)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (822)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (401)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Unit 1 Topic ICHDocument10 pagesUnit 1 Topic ICHZeyad A AbdullahNo ratings yet
- QA - Market Complaint Unit 4Document2 pagesQA - Market Complaint Unit 4Zeyad A AbdullahNo ratings yet
- Form 125 Example Batch Reconciliation Sheet For Tablet PackingDocument4 pagesForm 125 Example Batch Reconciliation Sheet For Tablet PackingZeyad A AbdullahNo ratings yet
- Pharmacognosy Unit 4 5th Short Important Questions Shahruddin KhanDocument16 pagesPharmacognosy Unit 4 5th Short Important Questions Shahruddin KhanZeyad A AbdullahNo ratings yet
- Form 160 Example - Line - Clearance Opening and Cleaning Form For Tablet PackingDocument8 pagesForm 160 Example - Line - Clearance Opening and Cleaning Form For Tablet PackingZeyad A AbdullahNo ratings yet
- Form 140 Visitor Entry Into The FactoryDocument1 pageForm 140 Visitor Entry Into The FactoryZeyad A AbdullahNo ratings yet
- U 1 PP 2 4th Sem B Pharm Pharmacy Wala Shahruddin Khan 18811494Document7 pagesU 1 PP 2 4th Sem B Pharm Pharmacy Wala Shahruddin Khan 18811494Zeyad A AbdullahNo ratings yet
- Form 085 Released StickersDocument1 pageForm 085 Released StickersZeyad A AbdullahNo ratings yet
- Form 400 Employee Signature RegisterDocument1 pageForm 400 Employee Signature RegisterZeyad A AbdullahNo ratings yet
- Vol. 3, Issue 3, March 2015, PharmaTutor, Paper-4Document7 pagesVol. 3, Issue 3, March 2015, PharmaTutor, Paper-4Zeyad A AbdullahNo ratings yet
- List of Glassware Used in Pharmaceuticals - PharmaguidelineDocument4 pagesList of Glassware Used in Pharmaceuticals - PharmaguidelineZeyad A AbdullahNo ratings yet
- Melatonin Oral DropDocument3 pagesMelatonin Oral DropZeyad A AbdullahNo ratings yet
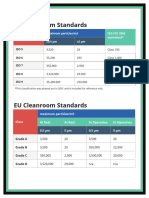
- Us Cleanroom Standards CompliancewireDocument1 pageUs Cleanroom Standards CompliancewireZeyad A AbdullahNo ratings yet
- RRT - DB - Mat WND enDocument2 pagesRRT - DB - Mat WND enZeyad A AbdullahNo ratings yet
- Project 1 7/5/2021Document1 pageProject 1 7/5/2021Zeyad A AbdullahNo ratings yet
- Catalogue Tablet-Press 2019 V3Document16 pagesCatalogue Tablet-Press 2019 V3Zeyad A AbdullahNo ratings yet
- PortaFab Mezzanine BrochureDocument2 pagesPortaFab Mezzanine BrochureZeyad A AbdullahNo ratings yet
- GMP Clearance GuidanceDocument84 pagesGMP Clearance GuidanceZeyad A AbdullahNo ratings yet
- Tube Filling and Sealing Machine Lab ModelDocument5 pagesTube Filling and Sealing Machine Lab ModelZeyad A AbdullahNo ratings yet
- IndJPhaEdRes 54 3s s473Document12 pagesIndJPhaEdRes 54 3s s473Zeyad A AbdullahNo ratings yet
- Ludipress Technical InformationDocument8 pagesLudipress Technical InformationZeyad A AbdullahNo ratings yet
- Product MonographDocument31 pagesProduct MonographZeyad A AbdullahNo ratings yet
- Tech Bulletin - Suglets - More Than MonographDocument2 pagesTech Bulletin - Suglets - More Than MonographZeyad A AbdullahNo ratings yet
- Sodium Stearyl Fumarate Excipient Pubchem 1559559385Document2 pagesSodium Stearyl Fumarate Excipient Pubchem 1559559385Zeyad A AbdullahNo ratings yet
- Crosslinked Povidone Pubchem 1482747992Document4 pagesCrosslinked Povidone Pubchem 1482747992Zeyad A AbdullahNo ratings yet
- S 065 LBLDocument10 pagesS 065 LBLZeyad A AbdullahNo ratings yet
- 09 Nov 2016 180959770P8GWIJNEANNEXUREDocument2 pages09 Nov 2016 180959770P8GWIJNEANNEXUREZeyad A AbdullahNo ratings yet
- 0Document1 page0Zeyad A AbdullahNo ratings yet
- What Is Environmental Monitoring in Pharmaceutical IndustryDocument12 pagesWhat Is Environmental Monitoring in Pharmaceutical IndustryZeyad A AbdullahNo ratings yet
- Good Practice Manufacturing Operation SampleDocument21 pagesGood Practice Manufacturing Operation SampleZeyad A AbdullahNo ratings yet