You might also like
- Synthesis of Well-Defined Allyl End-Functionalized Polystyrene by Atom Transfer Radical Polymerization With An Ally I Halide InitiatorDocument4 pagesSynthesis of Well-Defined Allyl End-Functionalized Polystyrene by Atom Transfer Radical Polymerization With An Ally I Halide InitiatorHeloisa Gabriele Miranda da SilvaNo ratings yet
- OJCV028I04P1815-1819 (J.AI - Percb 1)Document5 pagesOJCV028I04P1815-1819 (J.AI - Percb 1)Nur Aini IktikhafsariNo ratings yet
- Dlelectrlc Properties Electrolyte Solutions. 1. Sodium Iodide in Seven Solvents at Various TemperaturesDocument5 pagesDlelectrlc Properties Electrolyte Solutions. 1. Sodium Iodide in Seven Solvents at Various Temperaturesed roNo ratings yet
- The Cupric and Ferric Citrate ComplexesDocument9 pagesThe Cupric and Ferric Citrate ComplexesDemigodNo ratings yet
- Pharmaceutical Analysis: Mass SpectrometryDocument32 pagesPharmaceutical Analysis: Mass Spectrometryesie345No ratings yet
- Mo JAEDocument4 pagesMo JAEThanhThao TranNo ratings yet
- Galvanostatic Anodization of Titanium-Ii - Reactions Efficiencies and Electrochemical Behaviour ModelDocument9 pagesGalvanostatic Anodization of Titanium-Ii - Reactions Efficiencies and Electrochemical Behaviour Modelgundul123No ratings yet
- Practical Inorganic II EditedDocument163 pagesPractical Inorganic II EditedMosisa DugasaNo ratings yet
- CHM 161 Spectrophotometry: Analysis of Iron (II) in An Aqueous SolutionDocument10 pagesCHM 161 Spectrophotometry: Analysis of Iron (II) in An Aqueous SolutionPakistan ideologueNo ratings yet
- 1998 Nature Solid State DSSCDocument4 pages1998 Nature Solid State DSSCEka Cahya PrimaNo ratings yet
- Takahashi 1979Document6 pagesTakahashi 1979habbibrachmanNo ratings yet
- Rao-Chickerur1972 Article ComplexometricDeterminationOfLDocument1 pageRao-Chickerur1972 Article ComplexometricDeterminationOfLDavid ballena guerreroNo ratings yet
- Analytical ChemistryDocument7 pagesAnalytical Chemistrynagasri ponnalaNo ratings yet
- Fluorescencequenching of Rhodamine6Gin Methanol A T High ConcentrationDocument7 pagesFluorescencequenching of Rhodamine6Gin Methanol A T High Concentrationprakush_prakushNo ratings yet
- 1974 - Soga - Copolymerization of Carbon Dioxide With PropyleneimineDocument11 pages1974 - Soga - Copolymerization of Carbon Dioxide With PropyleneimineViraj EdirisingheNo ratings yet
- Awake!, Mar 2011Document4 pagesAwake!, Mar 2011emediageNo ratings yet
- Short Communications: Coal Gasification by Microwave Plasma in Water VapourDocument3 pagesShort Communications: Coal Gasification by Microwave Plasma in Water VapourDinda Naiya AzhariNo ratings yet
- Studies On The Solvent Extraction of Trivalent Lanthanides With Hexafluoroacetylacetone (HFAA) and Tri-N-Octylphosphineoxide (TOPO)Document6 pagesStudies On The Solvent Extraction of Trivalent Lanthanides With Hexafluoroacetylacetone (HFAA) and Tri-N-Octylphosphineoxide (TOPO)Ange QuintriqueoNo ratings yet
- Lab Manual Metal Acetylacetonate Complexes WebDocument22 pagesLab Manual Metal Acetylacetonate Complexes WebRahul GuptaNo ratings yet
- MG Ion Effect On Stability of Double-Stranded Polynucleotide Formed by Polyriboinosinic and Polyribocytidilic ChainsDocument9 pagesMG Ion Effect On Stability of Double-Stranded Polynucleotide Formed by Polyriboinosinic and Polyribocytidilic ChainsHagi DevNo ratings yet
- A Secondary Dithizone Complex Containing Both Silver and MercuryDocument10 pagesA Secondary Dithizone Complex Containing Both Silver and MercuryDiễn Đàn Hóa HọcNo ratings yet
- Photocatalytic Degradation of Methyl Orange by Polyoxometalates Supported On Yttrium-Doped TiO2Document6 pagesPhotocatalytic Degradation of Methyl Orange by Polyoxometalates Supported On Yttrium-Doped TiO2AstrialdelinaNo ratings yet
- The Main of This Research Was To Synthesize: Experimental Part Materials and MethodsDocument11 pagesThe Main of This Research Was To Synthesize: Experimental Part Materials and MethodsClaudiaDrăganNo ratings yet
- Instrumental Methods of AnalysisDocument10 pagesInstrumental Methods of AnalysisChemistry BNMITNo ratings yet
- Lab 3Document10 pagesLab 3Rahul Goel0% (1)
- Molecules-06-00831 PC FerDocument14 pagesMolecules-06-00831 PC FerPopusoi AnaNo ratings yet
- Novel Sol-Gel Method of Synthesis of Pure and Aluminum Doped Tio NanoparticlesDocument4 pagesNovel Sol-Gel Method of Synthesis of Pure and Aluminum Doped Tio NanoparticlesFahim ShahriarNo ratings yet
- Sensors: Determination of Trace Antimony (III) by Adsorption Voltammetry at Carbon Paste ElectrodeDocument9 pagesSensors: Determination of Trace Antimony (III) by Adsorption Voltammetry at Carbon Paste ElectrodeŞebnem İlarslanNo ratings yet
- 10.1038@s41929 018 0169 3Document8 pages10.1038@s41929 018 0169 3Patrik ConkaNo ratings yet
- Lab #11: Determination of A Chemical Equilibrium Constant: ObjectivesDocument0 pagesLab #11: Determination of A Chemical Equilibrium Constant: ObjectivesPablo BernalNo ratings yet
- Anatase Photo MB PHDocument5 pagesAnatase Photo MB PHNahed YacoubNo ratings yet
- Modified Photocatalyst P-Tio /PTDocument3 pagesModified Photocatalyst P-Tio /PTYulissa RamirezNo ratings yet
- Characterization of Chitosan. Influence of Ionic Strength and Degree of AcetylatiDocument5 pagesCharacterization of Chitosan. Influence of Ionic Strength and Degree of AcetylatiAlejandra Rojas-OlivosNo ratings yet
- J.Yang, W. H. Song, Y. Q. Ma, R. L. Zhang, and Y. P. SunDocument13 pagesJ.Yang, W. H. Song, Y. Q. Ma, R. L. Zhang, and Y. P. SunSruthi DevNo ratings yet
- Part 3 Using Light For DetectionDocument48 pagesPart 3 Using Light For DetectionJosé ClementeNo ratings yet
- Solubility Product of Hexavalent Uranium Hydrous Oxide: Original PaperDocument6 pagesSolubility Product of Hexavalent Uranium Hydrous Oxide: Original PaperKhairun NisaNo ratings yet
- Sensors: Effect of Tio On The Gas Sensing Features of Tio /pani NanocompositesDocument8 pagesSensors: Effect of Tio On The Gas Sensing Features of Tio /pani NanocompositesSarmad Sabih Al.obaidiNo ratings yet
- Thermodynamic Properties of Titanium Dioxide, Niobium Dioxide and Their Solid Solutions at High TemperatureDocument9 pagesThermodynamic Properties of Titanium Dioxide, Niobium Dioxide and Their Solid Solutions at High TemperatureNaufal AdityasNo ratings yet
- 01 Article - TC PolymeDocument13 pages01 Article - TC Polymeprint.15No ratings yet
- Full Paper: Photoacoustic Measurements of Porphyrin Triplet-State Quantum Yields and Singlet-Oxygen EfficienciesDocument9 pagesFull Paper: Photoacoustic Measurements of Porphyrin Triplet-State Quantum Yields and Singlet-Oxygen EfficienciesMariaNo ratings yet
- Infrared Spectra of Adsorbed Ethene On The Catalyst: Europt-1Document7 pagesInfrared Spectra of Adsorbed Ethene On The Catalyst: Europt-1María José Cortez RamírezNo ratings yet
- Research Article: Nitrogen-Doped Tio Nanotube Arrays With Enhanced Photoelectrochemical PropertyDocument8 pagesResearch Article: Nitrogen-Doped Tio Nanotube Arrays With Enhanced Photoelectrochemical PropertyJauhar FarrasyahNo ratings yet
- Experiment 10Document6 pagesExperiment 10Roman100% (2)
- Benson 1975, Comparaison Phtalayade Et FluorescamineDocument4 pagesBenson 1975, Comparaison Phtalayade Et Fluorescaminearnaud.fillaudeau44No ratings yet
- Articles: Structural Investigation of The Hydrolysis-Condensation Process of Modified Titanium IsopropoxideDocument5 pagesArticles: Structural Investigation of The Hydrolysis-Condensation Process of Modified Titanium IsopropoxideDeddy Triyono Nugroho AdiNo ratings yet
- (E) Unravelling The Mechanism of PhotoinducedDocument6 pages(E) Unravelling The Mechanism of PhotoinducedjvuszmpwthlfhbtqttNo ratings yet
- Lumino Cobre IIDocument7 pagesLumino Cobre IICalamidad Loco PeligroNo ratings yet
- 10.23 A Study On Adsorption Onto TODGA Resin After Electrolytic Reduction in ERIX Process For Reprocessing Spent FBR-MOX FuelDocument6 pages10.23 A Study On Adsorption Onto TODGA Resin After Electrolytic Reduction in ERIX Process For Reprocessing Spent FBR-MOX FuelPiter ColumboNo ratings yet
- Fybsc Lab Manual 2020-21 Sem II Che1203Document47 pagesFybsc Lab Manual 2020-21 Sem II Che1203Pratiksha GoreNo ratings yet
- UV SpectrosDocument25 pagesUV SpectrosokaciaNo ratings yet
- Hydration of Propylene Oxide by Hydrogen Peroxide Over Titania CatalystsDocument6 pagesHydration of Propylene Oxide by Hydrogen Peroxide Over Titania CatalystsA MahmoodNo ratings yet
- Synthesis of 1,4-Benzenedicarbonyl Thiourea Resins and Their Adsorption Properties For Ag (I)Document6 pagesSynthesis of 1,4-Benzenedicarbonyl Thiourea Resins and Their Adsorption Properties For Ag (I)Viviana TorresNo ratings yet
- 05 - Chapter 2 PDFDocument99 pages05 - Chapter 2 PDFUmme AbdullahNo ratings yet
- Experiment 2Document5 pagesExperiment 2VanandiNo ratings yet
- 3Document6 pages3Diego Alejandro Hurtado BalcazarNo ratings yet
- Evaluation of The Gas Law ConstantDocument14 pagesEvaluation of The Gas Law Constantjikhei100% (1)
- Sankalp Sheet - 1 Lecture 1: Atomic Structure (Discovery of Sub Atomic Particles & Waves)Document2 pagesSankalp Sheet - 1 Lecture 1: Atomic Structure (Discovery of Sub Atomic Particles & Waves)GcgNo ratings yet
- EdtaDocument13 pagesEdtaChongZY100% (1)
- XXIVth International Congress of Pure and Applied Chemistry: Plenary and Main Section Lectures Presented at Hamburg, Federal Republic of Germany, 2–8 September 1973From EverandXXIVth International Congress of Pure and Applied Chemistry: Plenary and Main Section Lectures Presented at Hamburg, Federal Republic of Germany, 2–8 September 1973No ratings yet
- Macromolecular Microsymposium — 16: Main Lectures Presented at the Sixteenth Microsymposium on Macromolecules (Advances in Scattering Methods), Prague, 12 - 16 July 1976From EverandMacromolecular Microsymposium — 16: Main Lectures Presented at the Sixteenth Microsymposium on Macromolecules (Advances in Scattering Methods), Prague, 12 - 16 July 1976B. SedláčekNo ratings yet
- HL Organic ChemistryDocument9 pagesHL Organic ChemistryMithran R TIPSNo ratings yet
- Calculus MSDocument14 pagesCalculus MSMithran R TIPSNo ratings yet
- Calculus WKDocument6 pagesCalculus WKMithran R TIPSNo ratings yet
- ME1 RevQnsDocument20 pagesME1 RevQnsMithran R TIPSNo ratings yet
- ME1 RevQns MSDocument18 pagesME1 RevQns MSMithran R TIPSNo ratings yet
- ICT Theory Rev WSDocument29 pagesICT Theory Rev WSMithran R TIPSNo ratings yet
- ICT Gr10 ME1 Theory Revn MSDocument9 pagesICT Gr10 ME1 Theory Revn MSMithran R TIPSNo ratings yet
- PIGMENTS AND POSTER PAINTS (Chemistry Project)Document29 pagesPIGMENTS AND POSTER PAINTS (Chemistry Project)Aryan GuptaNo ratings yet
- Guide To Best Practices in Ocean Alkalinity Enhancement ResearchDocument242 pagesGuide To Best Practices in Ocean Alkalinity Enhancement Researchqinglin1596433No ratings yet
- Sodium Carbonate: NA CODocument44 pagesSodium Carbonate: NA COMg HNo ratings yet
- Angle Gauges PDFDocument2 pagesAngle Gauges PDFpankajdharmadhikariNo ratings yet
- Preparation of Mohr's Salt ProjectDocument23 pagesPreparation of Mohr's Salt ProjectArjun Bajpai100% (1)
- Science6 q1 Mod1les3 Factors Affecting Solubility FINAL08032020Document19 pagesScience6 q1 Mod1les3 Factors Affecting Solubility FINAL08032020Gin CayobitNo ratings yet
- Carbon 94 (2015) 243-255 (Koh)Document13 pagesCarbon 94 (2015) 243-255 (Koh)Chuah Chong YangNo ratings yet
- Pinched Tube ReactorDocument8 pagesPinched Tube ReactorManoj BNo ratings yet
- 274-Article Text-1503-1-10-20230609Document8 pages274-Article Text-1503-1-10-20230609jalalNo ratings yet
- 1 - Exp4 - Dynaz Aqilah Binti Mohd SamsulDocument4 pages1 - Exp4 - Dynaz Aqilah Binti Mohd SamsulDynazze 04No ratings yet
- Reference Electrodes With Salt Bridges Contained in NanoporousDocument7 pagesReference Electrodes With Salt Bridges Contained in NanoporousSahana SarkarNo ratings yet
- Fluorescent Magnetic TestingDocument29 pagesFluorescent Magnetic TestingAlzaki Abdullah100% (1)
- pw1 PDFDocument10 pagespw1 PDFUlvi AgayevNo ratings yet
- 1 s2.0 S0021967308005104 MainDocument8 pages1 s2.0 S0021967308005104 MainYến NhiNo ratings yet
- Coa Lecitina de Soya Alimentos Lote A-074-20Document1 pageCoa Lecitina de Soya Alimentos Lote A-074-20Carlos SiuNo ratings yet
- Mass Transfer Operations 2020Document325 pagesMass Transfer Operations 2020EJ TanNo ratings yet
- MM-18 - Bilge Separator - OPERATION MANUALDocument24 pagesMM-18 - Bilge Separator - OPERATION MANUALKyaw Swar Latt100% (2)
- Work Out Chemistry GCSE (PDFDrive)Document163 pagesWork Out Chemistry GCSE (PDFDrive)Rico ChanNo ratings yet
- Carbohydrates TestsDocument3 pagesCarbohydrates TestsKings PrideNo ratings yet
- GECC 1 Proven Corrosion PreventionDocument3 pagesGECC 1 Proven Corrosion PreventionAchmad Arifudin HidayatullohNo ratings yet
- Sugar Cane Fertilizer Chart For Coming YearDocument2 pagesSugar Cane Fertilizer Chart For Coming YearAmeet Kudche100% (1)
- Amie Syllabus Sec B ChemicalDocument6 pagesAmie Syllabus Sec B ChemicalArunkumarNo ratings yet
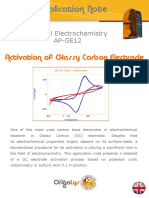
- AP GE12 Glassy Carbon ActivationDocument6 pagesAP GE12 Glassy Carbon ActivationMatthieu EmondNo ratings yet
- HT Mobilith SHC 460 EngDocument2 pagesHT Mobilith SHC 460 Engaxd123No ratings yet
- Astm G 35 - 98 - RZM1LTK4Document3 pagesAstm G 35 - 98 - RZM1LTK4Cordova RaphaelNo ratings yet
- NATURAL DYES and DYEINGDocument2 pagesNATURAL DYES and DYEINGHomer RendonNo ratings yet
- Comparison of The Concentration of Phenolic Constituents SugarcaneDocument7 pagesComparison of The Concentration of Phenolic Constituents SugarcaneMolly0630No ratings yet
- (Some Relevant Equations Given Below) : P-FunctionsDocument2 pages(Some Relevant Equations Given Below) : P-FunctionsKrishan PrajapatiNo ratings yet
- Lignin: DR 900 Analytical ProcedureDocument4 pagesLignin: DR 900 Analytical Procedurewulalan wulanNo ratings yet
- Welding ReportDocument3 pagesWelding ReportChanten NanNo ratings yet