You might also like
- Advances in Cell and Molecular DiagnosticsFrom EverandAdvances in Cell and Molecular DiagnosticsRating: 5 out of 5 stars5/5 (1)
- Genomic Data Sharing: Case Studies, Challenges, and Opportunities for Precision MedicineFrom EverandGenomic Data Sharing: Case Studies, Challenges, and Opportunities for Precision MedicineJennifer B. MccormickNo ratings yet
- Essentials of Genomic and Personalized MedicineFrom EverandEssentials of Genomic and Personalized MedicineRating: 2 out of 5 stars2/5 (1)
- Integration and Visualization of Gene Selection and Gene Regulatory Networks for Cancer GenomeFrom EverandIntegration and Visualization of Gene Selection and Gene Regulatory Networks for Cancer GenomeNo ratings yet
- Managing Health in the Genomic Era: A Guide to Family Health History and Disease RiskFrom EverandManaging Health in the Genomic Era: A Guide to Family Health History and Disease RiskNo ratings yet
- Popdx: An Automated Framework For Patient Phenotyping Across 392 246 Individuals in The Uk Biobank StudyDocument11 pagesPopdx: An Automated Framework For Patient Phenotyping Across 392 246 Individuals in The Uk Biobank StudysubaNo ratings yet
- Building A Knowledge Graph To Enable Precision Medicine: Data DescriptorDocument16 pagesBuilding A Knowledge Graph To Enable Precision Medicine: Data DescriptorPablo Ernesto Vigneaux WiltonNo ratings yet
- Clustering Rare Diseases Within An Ontology-Enriched Knowledge GraphDocument24 pagesClustering Rare Diseases Within An Ontology-Enriched Knowledge GraphPablo Ernesto Vigneaux WiltonNo ratings yet
- ICT583 Assignment 1Document4 pagesICT583 Assignment 1Salman CheemaNo ratings yet
- Leveraging Biomedical and Healthcare Data: Semantics, Analytics and KnowledgeFrom EverandLeveraging Biomedical and Healthcare Data: Semantics, Analytics and KnowledgeFiras KobeissyNo ratings yet
- Between the Lines of Genetic Code: Genetic Interactions in Understanding Disease and Complex PhenotypesFrom EverandBetween the Lines of Genetic Code: Genetic Interactions in Understanding Disease and Complex PhenotypesLeonid PadyukovNo ratings yet
- Neurocritical Care Informatics: Translating Raw Data into Bedside ActionFrom EverandNeurocritical Care Informatics: Translating Raw Data into Bedside ActionMichael De GeorgiaNo ratings yet
- Ophthalmic Artery Doppler in The Complementary Diagnosis of Preeclampsia: A Systematic Review and Meta-AnalysisDocument11 pagesOphthalmic Artery Doppler in The Complementary Diagnosis of Preeclampsia: A Systematic Review and Meta-AnalysisValia ChuchonNo ratings yet
- Genomic Medicine: A Practical GuideFrom EverandGenomic Medicine: A Practical GuideLaura J. TafeNo ratings yet
- Biomarkers and Biosensors: Detection and Binding to Biosensor Surfaces and Biomarkers ApplicationsFrom EverandBiomarkers and Biosensors: Detection and Binding to Biosensor Surfaces and Biomarkers ApplicationsNo ratings yet
- 2023 Peripheral Neuropathy in Older People Is Associated With ReducedDocument8 pages2023 Peripheral Neuropathy in Older People Is Associated With ReducedElisabeth Permatasari SidabutarNo ratings yet
- Brucella Melitensis: Identification and Characterization of Potential Drug TargetsFrom EverandBrucella Melitensis: Identification and Characterization of Potential Drug TargetsJangampalli Adi PradeepkiranNo ratings yet
- Genomics of Rare Diseases: Understanding Disease Genetics Using Genomic ApproachesFrom EverandGenomics of Rare Diseases: Understanding Disease Genetics Using Genomic ApproachesClaudia Gonzaga-JaureguiNo ratings yet
- Clinical Ethics at the Crossroads of Genetic and Reproductive TechnologiesFrom EverandClinical Ethics at the Crossroads of Genetic and Reproductive TechnologiesSorin HostiucNo ratings yet
- Life Histories of Genetic Disease: Patterns and Prevention in Postwar Medical GeneticsFrom EverandLife Histories of Genetic Disease: Patterns and Prevention in Postwar Medical GeneticsNo ratings yet
- 2022 02 28 22271643v1 FullDocument49 pages2022 02 28 22271643v1 FullA GouveiaNo ratings yet
- Investigating Associations Between Blood Metabolites, Later Life Brain Imaging Measures, and Genetic Risk For Alzheimer's DiseaseDocument13 pagesInvestigating Associations Between Blood Metabolites, Later Life Brain Imaging Measures, and Genetic Risk For Alzheimer's DiseaseCamila Alejandra Inostroza RiveraNo ratings yet
- Preprint-2023 05 14 23289955 FullDocument23 pagesPreprint-2023 05 14 23289955 Fullben-zhuNo ratings yet
- 2022 12 07 22283238v1 FullDocument55 pages2022 12 07 22283238v1 FullKarl GemayelNo ratings yet
- Finding Novel Molecular Connections Between Developmental Processes and DiseaseDocument12 pagesFinding Novel Molecular Connections Between Developmental Processes and DiseasezottisadNo ratings yet
- Pathway Analysis for Drug Discovery: Computational Infrastructure and ApplicationsFrom EverandPathway Analysis for Drug Discovery: Computational Infrastructure and ApplicationsNo ratings yet
- A Genetics-First Approach To Dissecting The Heterogeneity of Autism Phenotypic Comparison of Autism Risk Copy Number VariantsDocument27 pagesA Genetics-First Approach To Dissecting The Heterogeneity of Autism Phenotypic Comparison of Autism Risk Copy Number VariantsTiago ChavesNo ratings yet
- Nursing InformaticsDocument3 pagesNursing InformaticsGabriel Luis SolanoNo ratings yet
- Artificial Intelligence for Medicine: An Applied Reference for Methods and ApplicationsFrom EverandArtificial Intelligence for Medicine: An Applied Reference for Methods and ApplicationsShai Ben- DavidNo ratings yet
- Clinical Applications for Next-Generation SequencingFrom EverandClinical Applications for Next-Generation SequencingUrszula DemkowRating: 4 out of 5 stars4/5 (2)
- 2021 06 29 21254249 FullDocument28 pages2021 06 29 21254249 FullBharat TradersNo ratings yet
- Emery and Rimoin’s Principles and Practice of Medical Genetics and Genomics: Clinical Principles and ApplicationsFrom EverandEmery and Rimoin’s Principles and Practice of Medical Genetics and Genomics: Clinical Principles and ApplicationsReed E. PyeritzNo ratings yet
- 2016 Book SecondaryAnalysisOfElectronicH PDFDocument435 pages2016 Book SecondaryAnalysisOfElectronicH PDFSharon DhillonNo ratings yet
- 2022 03 23 22272776 FullDocument21 pages2022 03 23 22272776 Fullsg.gis279No ratings yet
- Modern Inference Based on Health-Related Markers: Biomarkers and Statistical Decision MakingFrom EverandModern Inference Based on Health-Related Markers: Biomarkers and Statistical Decision MakingAlbert VexlerNo ratings yet
- International Journal of Medical InformaticsDocument9 pagesInternational Journal of Medical InformaticsLetícia RosaNo ratings yet
- V71I12 - A Breakthrough in Schizophrenia GeneticsDocument2 pagesV71I12 - A Breakthrough in Schizophrenia GeneticsOscar Alejandro Cardenas QuinteroNo ratings yet
- Pope - Moseley@cpr - Ku.dk: To The EditorDocument2 pagesPope - Moseley@cpr - Ku.dk: To The EditorLili MorgadoNo ratings yet
- Self-assessment Questions for Clinical Molecular GeneticsFrom EverandSelf-assessment Questions for Clinical Molecular GeneticsRating: 5 out of 5 stars5/5 (1)
- Precision Medicine Oncology: A PrimerFrom EverandPrecision Medicine Oncology: A PrimerLorna Rodriguez-RodriguezNo ratings yet
- The Dynamics of Biomarkers Across The Clinical Spectrum of ADDocument16 pagesThe Dynamics of Biomarkers Across The Clinical Spectrum of ADBrenda GonçalvesNo ratings yet
- Nu716 m7 - Data Analytics Part IIDocument6 pagesNu716 m7 - Data Analytics Part IIapi-577441503No ratings yet
- Risk Assessment POD2023Document10 pagesRisk Assessment POD2023Cláudia Regina FernandesNo ratings yet
- Emery and Rimoin’s Principles and Practice of Medical Genetics and Genomics: FoundationsFrom EverandEmery and Rimoin’s Principles and Practice of Medical Genetics and Genomics: FoundationsReed E. PyeritzNo ratings yet
- Jurnal Internasional 3Document4 pagesJurnal Internasional 3Putri UtamiNo ratings yet
- Precision Medicine in Cancer TherapyFrom EverandPrecision Medicine in Cancer TherapyDaniel D. Von HoffNo ratings yet
- Genomic and Precision Medicine: Infectious and Inflammatory DiseaseFrom EverandGenomic and Precision Medicine: Infectious and Inflammatory DiseaseNo ratings yet
- Genes Parents NeurodelevopDocument33 pagesGenes Parents NeurodelevopDa CuNo ratings yet
- 10 1016@j Eclinm 2019 05 003Document7 pages10 1016@j Eclinm 2019 05 003Hijaz HijaNo ratings yet
- Sensors: Comparison of Machine Learning Algorithms in The Prediction of Hospitalized Patients With SchizophreniaDocument15 pagesSensors: Comparison of Machine Learning Algorithms in The Prediction of Hospitalized Patients With SchizophreniaMs. S.Sridevi Vels UniversityNo ratings yet
- For CorrectionDocument8 pagesFor CorrectionAvijit ChaudhuriNo ratings yet
- Functional Genomics in HCC 2005Document6 pagesFunctional Genomics in HCC 2005johnyap11No ratings yet
- Modeling and Analyzing Predictive Monthly Survival in Females Diagnosed With Gynecological CancersDocument10 pagesModeling and Analyzing Predictive Monthly Survival in Females Diagnosed With Gynecological CancersIJPHSNo ratings yet
- Discovering Comorbid Diseases Using An Inter-DiseaDocument10 pagesDiscovering Comorbid Diseases Using An Inter-DiseaNealNo ratings yet
- Clinical Aspects of Multiple Sclerosis Essentials and Current UpdatesFrom EverandClinical Aspects of Multiple Sclerosis Essentials and Current UpdatesShitiz SriwastavaNo ratings yet
- Challenges in Delivery of Therapeutic Genomics and ProteomicsFrom EverandChallenges in Delivery of Therapeutic Genomics and ProteomicsAmbikanandan MisraNo ratings yet
- Final Term Paper Draft 2Document33 pagesFinal Term Paper Draft 2Soham DattaNo ratings yet
- J. Sil 1Document6 pagesJ. Sil 1Soham DattaNo ratings yet
- Deep Recurrent Q Reinforcement Learning Model To PDocument10 pagesDeep Recurrent Q Reinforcement Learning Model To PSoham DattaNo ratings yet
- DRL NewDocument12 pagesDRL NewSoham DattaNo ratings yet
- Hashing Important TheoremsDocument26 pagesHashing Important TheoremsSoham DattaNo ratings yet
- NeuralDocument33 pagesNeuralSoham DattaNo ratings yet
- The Promise of Convolutional Neural NetworksDocument13 pagesThe Promise of Convolutional Neural NetworksSoham DattaNo ratings yet
- Common Record Sheet ModifiedDocument5 pagesCommon Record Sheet ModifiedSoham DattaNo ratings yet
- 2023csm007 DBMS 1Document6 pages2023csm007 DBMS 1Soham DattaNo ratings yet
- Inter Hospital Alzheimer's Disease Detection CNNDocument12 pagesInter Hospital Alzheimer's Disease Detection CNNSoham DattaNo ratings yet
- ETRI Journal - 2024 - Bang - Alzheimer S Disease Recognition From Spontaneous Speech Using Large Language ModelsDocument10 pagesETRI Journal - 2024 - Bang - Alzheimer S Disease Recognition From Spontaneous Speech Using Large Language ModelsSoham DattaNo ratings yet
- Multimodal Deep Learning ApproachDocument11 pagesMultimodal Deep Learning ApproachSoham DattaNo ratings yet
- Souk Menu 2022Document8 pagesSouk Menu 2022Soham DattaNo ratings yet
- CMINDS AdmissionTest - SYLLABUS Apr22Document4 pagesCMINDS AdmissionTest - SYLLABUS Apr22Soham DattaNo ratings yet
- Eigenvalues and EigenvectorsDocument33 pagesEigenvalues and EigenvectorsSoham DattaNo ratings yet
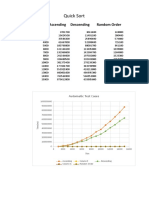
- Quick Sort: Size Ascending Descending Random OrderDocument5 pagesQuick Sort: Size Ascending Descending Random OrderSoham DattaNo ratings yet
- Da Jo: Auareccurrence JoeeDocument5 pagesDa Jo: Auareccurrence JoeeSoham DattaNo ratings yet
- Project Programs FinalDocument16 pagesProject Programs FinalSoham DattaNo ratings yet
- Disadvantage of Cash FlowDocument29 pagesDisadvantage of Cash FlowSoham DattaNo ratings yet
- DartonismDocument4 pagesDartonismSoham DattaNo ratings yet
- Anirban Mukherjee Resume.Document2 pagesAnirban Mukherjee Resume.Soham DattaNo ratings yet
- CommerceDocument25 pagesCommerceSoham DattaNo ratings yet
- NCP GRAND CASE PRE C Nursing ProblemsDocument9 pagesNCP GRAND CASE PRE C Nursing ProblemsAngie Mandeoya100% (1)
- Disease and Disease TransmissionDocument48 pagesDisease and Disease TransmissionAYO NELSONNo ratings yet
- Heart DiseaseDocument40 pagesHeart DiseaseAhmer TahirNo ratings yet
- How We Conquered SmallpoxDocument4 pagesHow We Conquered SmallpoxNadir BaghdadNo ratings yet
- Dilution Reconstitution Injection GuideDocument2 pagesDilution Reconstitution Injection GuideBorislavaSavova100% (1)
- The Role of Acute Phase Proteins in Diagnosis and ManagementDocument6 pagesThe Role of Acute Phase Proteins in Diagnosis and ManagementCabinet VeterinarNo ratings yet
- White Tongue Causes & 10 Natural Treatments For White Tongue - Dr. AxeDocument10 pagesWhite Tongue Causes & 10 Natural Treatments For White Tongue - Dr. AxeRaghu100% (1)
- Fenotipos Bioquímicos y Fagotipos de Cepas de Salmonella Enteritidis Aisladas en Antofagasta, 1997-2000Document9 pagesFenotipos Bioquímicos y Fagotipos de Cepas de Salmonella Enteritidis Aisladas en Antofagasta, 1997-2000axel vargasNo ratings yet
- Nnyy3999 PDFDocument2 pagesNnyy3999 PDFAshwin SagarNo ratings yet
- Imaging of Solitary and Multiple Pulmonary NodulesDocument75 pagesImaging of Solitary and Multiple Pulmonary NodulesAnshulVarshneyNo ratings yet
- Bells PalsyDocument2 pagesBells PalsyJhevilin RMNo ratings yet
- Dead Space ManagementDocument26 pagesDead Space ManagementIgnasNo ratings yet
- AsplDocument9 pagesAsplCristinaNo ratings yet
- A Special Interview With Dr. Meryl Nass by Dr. Joseph MercolaDocument9 pagesA Special Interview With Dr. Meryl Nass by Dr. Joseph MercolaJonathan Robert Kraus (OutofMudProductions)No ratings yet
- Hospital MarketingDocument10 pagesHospital MarketingYogesh Parchani100% (1)
- GUIAS UCI para La Alimentación de Paciente CriticoDocument33 pagesGUIAS UCI para La Alimentación de Paciente CriticoFredy UmbarilaNo ratings yet
- LIVER CHIRROSIS and GERED QUESTION AND ANSWERSDocument10 pagesLIVER CHIRROSIS and GERED QUESTION AND ANSWERSjess_nookieNo ratings yet
- Renr Practice Test 9 FinalDocument12 pagesRenr Practice Test 9 FinalTk100% (2)
- MJMHS Jan 2019Document7 pagesMJMHS Jan 2019Sekretariat HemodialisisNo ratings yet
- Nursing Care Plan FinalDocument2 pagesNursing Care Plan FinalRy LlanesNo ratings yet
- COMMUNITY CASE STUDY Local Health Program of Regional UniversityDocument20 pagesCOMMUNITY CASE STUDY Local Health Program of Regional Universitymarlou agananNo ratings yet
- TWSTRS Severity ScaleDocument2 pagesTWSTRS Severity ScaleErri PratamaNo ratings yet
- Axis I Clinical Psychiatric Disorders - LMM-1Document11 pagesAxis I Clinical Psychiatric Disorders - LMM-1richhuangNo ratings yet
- Aplastic Anemia: Savita HandayaniDocument24 pagesAplastic Anemia: Savita HandayaniAnna Puteri GozaliNo ratings yet
- Grammar and VocabularyDocument2 pagesGrammar and Vocabularythiên bình trầnNo ratings yet
- Seminar ImnciDocument15 pagesSeminar Imnciseema devassy100% (1)
- Flow Cytometry,: Applications in TMDocument72 pagesFlow Cytometry,: Applications in TMMohandoss MurugesanNo ratings yet
- Ortho Referral Quick Reference SheetDocument2 pagesOrtho Referral Quick Reference SheetAbdelhamid FaragNo ratings yet
- SB 3 Flu VaccineDocument2 pagesSB 3 Flu VaccineBIANNE KATRINA GASAGASNo ratings yet
- ProQuestDocuments 2023 04 26Document3 pagesProQuestDocuments 2023 04 26Adriana Salazar MonjaNo ratings yet