You might also like
- Auto-Inflammatory Syndromes: Pathophysiology, Diagnosis, and ManagementFrom EverandAuto-Inflammatory Syndromes: Pathophysiology, Diagnosis, and ManagementPetros EfthimiouNo ratings yet
- Genetics of Endocrinology: Amit R. Majithia - David Altshuler - Joel N. HirschhornDocument20 pagesGenetics of Endocrinology: Amit R. Majithia - David Altshuler - Joel N. HirschhornDr Mehul Kumar ChourasiaNo ratings yet
- 2013 Clinical WES Nejmoa1306555Document10 pages2013 Clinical WES Nejmoa1306555MCuk2606No ratings yet
- Fgf13 Causes Synaptic Excitatory-Inhibitory: Disruption of Imbalance and Genetic Epilepsy and Febrile Seizures PlusDocument16 pagesFgf13 Causes Synaptic Excitatory-Inhibitory: Disruption of Imbalance and Genetic Epilepsy and Febrile Seizures Plushidayatul rahmoNo ratings yet
- Variant of TYR and Autoimmunity Susceptibility Loci in Generalized VitiligoDocument12 pagesVariant of TYR and Autoimmunity Susceptibility Loci in Generalized VitiligoIvo AfianiNo ratings yet
- The Disruption of Profiling of Serotonergic Neurons, Results in Autism-Related BehaviorsDocument22 pagesThe Disruption of Profiling of Serotonergic Neurons, Results in Autism-Related BehaviorsAngelinni Taglioni StangeNo ratings yet
- Nej Mo A 1516767Document11 pagesNej Mo A 1516767mayrapp16No ratings yet
- Pleiotropy in Complex Traits: Challenges and StrategiesDocument13 pagesPleiotropy in Complex Traits: Challenges and Strategiesthe invincibleNo ratings yet
- Foundations, Promises and Uncertainties of Personalized MedicineDocument7 pagesFoundations, Promises and Uncertainties of Personalized MedicineVeronica BalanNo ratings yet
- Charcot Marie Tooth DiseaseDocument9 pagesCharcot Marie Tooth DiseaseMihai CebotariNo ratings yet
- From Mendel To The Human Genome Project: The Implications For Nurse EducationDocument6 pagesFrom Mendel To The Human Genome Project: The Implications For Nurse EducationKome ČemuNo ratings yet
- Kotlyar 2020Document16 pagesKotlyar 2020Merab KvintradzeNo ratings yet
- Lehmann Werman 2016Document9 pagesLehmann Werman 2016Felipe MNo ratings yet
- Germline Mutations in Predisposition Genes in Pediatric CancerDocument11 pagesGermline Mutations in Predisposition Genes in Pediatric CancerGuadalupeA.OsorioNo ratings yet
- Fced GenetikDocument33 pagesFced GenetikyosuanugrahapratamaNo ratings yet
- E472 FullDocument17 pagesE472 FullC_DanteNo ratings yet
- A Haplotype Map of The Human GenomeDocument57 pagesA Haplotype Map of The Human GenomeJanina PuenteNo ratings yet
- Part 16 - Genes, The Environment, and DiseaseDocument226 pagesPart 16 - Genes, The Environment, and Diseasenklinh.y2021No ratings yet
- ALSDocument14 pagesALSZé CunhaNo ratings yet
- Sudden Infant Death Syndrome: Case-Control Frequency Differences at Genes Pertinent To Early Autonomic Nervous System Embryologic DevelopmentDocument5 pagesSudden Infant Death Syndrome: Case-Control Frequency Differences at Genes Pertinent To Early Autonomic Nervous System Embryologic DevelopmentJailyn Carrasquel HerreraNo ratings yet
- EditorialDocument2 pagesEditorialsebastoto778No ratings yet
- 1 Biomarkers Ruggeri Et Al 2013Document16 pages1 Biomarkers Ruggeri Et Al 2013sri309No ratings yet
- 33 PDFDocument4 pages33 PDFThelma CastlleNo ratings yet
- Greenberg1996 PDFDocument8 pagesGreenberg1996 PDFAraNo ratings yet
- Genetic Association Studies: Design, Analysis and InterpretationDocument8 pagesGenetic Association Studies: Design, Analysis and InterpretationfaizahNo ratings yet
- Panel1 GenomicMedicineAnUpdatedPrimer PDFDocument11 pagesPanel1 GenomicMedicineAnUpdatedPrimer PDFMariana ÁlvarezNo ratings yet
- Role For Hedgehog Signaling in Cranial-Suture Development and ObesityDocument9 pagesRole For Hedgehog Signaling in Cranial-Suture Development and ObesityDanny Alvarez FocacciNo ratings yet
- Pkan NejmDocument8 pagesPkan Nejmmartin.ruda93No ratings yet
- How To Interpret A GWASDocument10 pagesHow To Interpret A GWAStpatel0986No ratings yet
- Gene Hunting: J. PongraczDocument15 pagesGene Hunting: J. PongracztahirnaeemkhanNo ratings yet
- This Content Downloaded From 120.126.70.24 On Wed, 29 Mar 2023 11:59:10 UTCDocument8 pagesThis Content Downloaded From 120.126.70.24 On Wed, 29 Mar 2023 11:59:10 UTCmarcia suNo ratings yet
- 1 s2.0 S0888754321002251 MainDocument11 pages1 s2.0 S0888754321002251 Mainhilay.koseNo ratings yet
- Nrneurol 2011 65Document2 pagesNrneurol 2011 65lauravshNo ratings yet
- 1 s2.0 S2589004221008932 MainDocument11 pages1 s2.0 S2589004221008932 MainBen DresimNo ratings yet
- Artigo Cientifico Charcot Marie ToothDocument8 pagesArtigo Cientifico Charcot Marie ToothCleverson Da Silva MattosNo ratings yet
- Primer: Down SyndromeDocument20 pagesPrimer: Down SyndromeangelinaNo ratings yet
- Forward (2017)Document13 pagesForward (2017)DanieleRibeiroNo ratings yet
- ElderlyDocument1 pageElderlyapi-26340035No ratings yet
- Genomewide Scan of Hoarding in Sib Pairs in Which Both Sibs Have Gilles de La Tourette SyndromeDocument9 pagesGenomewide Scan of Hoarding in Sib Pairs in Which Both Sibs Have Gilles de La Tourette SyndromeCita Tri KusumaNo ratings yet
- Mutations in The MORC2 Gene Cause AxonalDocument11 pagesMutations in The MORC2 Gene Cause AxonalMILON KUMAR HORENo ratings yet
- Family History of Alzheimer's Disease and Cortical Thickness in Patients With DementiaDocument7 pagesFamily History of Alzheimer's Disease and Cortical Thickness in Patients With DementiaMarcos Josue Costa DiasNo ratings yet
- Gomez Ospina2016Document11 pagesGomez Ospina2016christian roblesNo ratings yet
- Progressive Increase of The Mutated Mitochondria1 DNA Fraction in Kearns-Sayre SyndromeDocument6 pagesProgressive Increase of The Mutated Mitochondria1 DNA Fraction in Kearns-Sayre SyndromeGréta BotyánszkiNo ratings yet
- Zheng 2013Document6 pagesZheng 2013namiraNo ratings yet
- Principles of Pharmacogenetics-Implications For The AnaesthetistDocument11 pagesPrinciples of Pharmacogenetics-Implications For The AnaesthetistLulu SetiyabudiNo ratings yet
- Female Predominance in Intermittent Exotropia: ConclusionsDocument2 pagesFemale Predominance in Intermittent Exotropia: ConclusionsIkmal ShahromNo ratings yet
- 2020 - Autism Genetics. Over 100 Risk Genes and Counting - ForrestDocument1 page2020 - Autism Genetics. Over 100 Risk Genes and Counting - ForrestJesus RiveraNo ratings yet
- Clinical Interpretation and Management of Genetic VariantsDocument14 pagesClinical Interpretation and Management of Genetic VariantsAnastasija ZuravlovaNo ratings yet
- Current Concepts Review: Ph. Debeer, L. de Smet, W. J. M. Van de Ven, G. Fabry, J.-P. FrynsDocument12 pagesCurrent Concepts Review: Ph. Debeer, L. de Smet, W. J. M. Van de Ven, G. Fabry, J.-P. FrynsDr. Pedro Javier Cadena GonzálezNo ratings yet
- Autismo EstudioDocument41 pagesAutismo Estudiomauricio lopezNo ratings yet
- Zheng Hong 郑 红 Department of Medical Genetics & Cell BiologyDocument63 pagesZheng Hong 郑 红 Department of Medical Genetics & Cell BiologyinakiNo ratings yet
- Brochure Genetics-The Basic FactsDocument7 pagesBrochure Genetics-The Basic FactsVale FlociaNo ratings yet
- Genomewide Association Study of Leprosy: Original ArticleDocument10 pagesGenomewide Association Study of Leprosy: Original ArticleFickry AdiansyahNo ratings yet
- 5 Ensuring Equity Diversity and Inclusiveness in Genetic Analysis Will Empower The Future ofDocument4 pages5 Ensuring Equity Diversity and Inclusiveness in Genetic Analysis Will Empower The Future ofabdeali hazariNo ratings yet
- Guimier Et Al. (2015) PMID 26437028 - 0Document14 pagesGuimier Et Al. (2015) PMID 26437028 - 0Filip MilošićNo ratings yet
- Background: The Genetic of AnticipationDocument3 pagesBackground: The Genetic of AnticipationAh LynNo ratings yet
- Melkersson Rosenthal Sindrome Reporte Caso InglesDocument4 pagesMelkersson Rosenthal Sindrome Reporte Caso InglesOscar Cabezas CalderónNo ratings yet
- Genetics 0727Document9 pagesGenetics 0727Implant Surgical GuidesNo ratings yet
- Journal Pre-Proof: Biological PsychiatryDocument38 pagesJournal Pre-Proof: Biological PsychiatryTakelooNo ratings yet
- Genome Sequencing in The ClinicDocument17 pagesGenome Sequencing in The ClinicChristopher NorrieNo ratings yet
- 5 Scherloc SlikaDocument1 page5 Scherloc SlikaMCuk2606No ratings yet
- 3 Scherloc SlikaDocument1 page3 Scherloc SlikaMCuk2606No ratings yet
- 2 Scherloc SlikaDocument1 page2 Scherloc SlikaMCuk2606No ratings yet
- Alfa 1 Antitripsin Manjak Review 2020 10.1056@NEJMra1910234Document13 pagesAlfa 1 Antitripsin Manjak Review 2020 10.1056@NEJMra1910234MCuk2606No ratings yet
- CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-ThalassemiaDocument9 pagesCRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-ThalassemiadrsourabhsinghNo ratings yet
- Ga1 Outcome Treatment Kevine Strauss 2020Document16 pagesGa1 Outcome Treatment Kevine Strauss 2020MCuk2606No ratings yet
- 647-Article Text-964-1-10-20200103Document4 pages647-Article Text-964-1-10-20200103MCuk2606No ratings yet
- 647-Article Text-964-1-10-20200103Document4 pages647-Article Text-964-1-10-20200103MCuk2606No ratings yet
- Biological Induced CorrosionDocument22 pagesBiological Induced CorrosionHemanth100% (1)
- A New Species of Riverine Crab of The Genus Sundathelphusa Bott, 1969 (Crustacea: Brachyura: Gecarcinucidae) From Northeastern Luzon, PhilippinesDocument10 pagesA New Species of Riverine Crab of The Genus Sundathelphusa Bott, 1969 (Crustacea: Brachyura: Gecarcinucidae) From Northeastern Luzon, PhilippinesNel YagosNo ratings yet
- BIO 328 Full Semester PackageDocument25 pagesBIO 328 Full Semester PackageNerdy Notes Inc.No ratings yet
- Syllabus CompletionDocument1 pageSyllabus CompletiongopimicroNo ratings yet
- Study Guide Cardio Tayang by Gextha 30 Maret 2015Document82 pagesStudy Guide Cardio Tayang by Gextha 30 Maret 2015Adi ParamarthaNo ratings yet
- Pulse Flour From Wheat MillerDocument23 pagesPulse Flour From Wheat MillerCao Trọng HiếuNo ratings yet
- ZIZKA Bromeliaceae ChileDocument22 pagesZIZKA Bromeliaceae ChileJoaquín Eduardo Sepúlveda AstudilloNo ratings yet
- Angiogenesis and Direct Myocardial RevascularizationDocument364 pagesAngiogenesis and Direct Myocardial RevascularizationPerdana SidaurukNo ratings yet
- CAPE Bio Mark SchemeDocument4 pagesCAPE Bio Mark Schemeron97150% (2)
- EASL 2021 Version 4 NewDocument691 pagesEASL 2021 Version 4 NewGupse Köroğlu AdalıNo ratings yet
- Personal Development Lesson 2Document54 pagesPersonal Development Lesson 2Ysay FranciscoNo ratings yet
- RPT SC Year 2 (DLP) 2022-2023Document21 pagesRPT SC Year 2 (DLP) 2022-2023ANNIE MARGARET A/P ANTHONY MoeNo ratings yet
- The Differences Between Asexual and Sexual ReproductionDocument2 pagesThe Differences Between Asexual and Sexual ReproductionAmirul IkhwanNo ratings yet
- Chapter 1Document4 pagesChapter 1Nikoleta RudnikNo ratings yet
- Gender and PsychotherapyDocument12 pagesGender and PsychotherapyEhh EnNaNo ratings yet
- Kimia Daun PandanDocument4 pagesKimia Daun Pandanlutfi_alhayathullahNo ratings yet
- OB Ultrasound Report Template 2Document1 pageOB Ultrasound Report Template 2PriyankaNo ratings yet
- Selina Solutions For Class 9 Biology Chapter 2 Cell The Unit of LifeDocument15 pagesSelina Solutions For Class 9 Biology Chapter 2 Cell The Unit of LifePrashun PriyadarshiNo ratings yet
- 7 Pearson SquareDocument19 pages7 Pearson Squareapi-235944649No ratings yet
- Sri Roth 2000Document11 pagesSri Roth 2000ottoojuniiorNo ratings yet
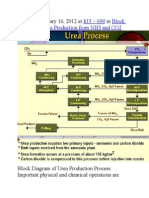
- Published January 16, 2012 at In: 813 × 699 Block Diagram of Urea Production From NH3 and CO2Document9 pagesPublished January 16, 2012 at In: 813 × 699 Block Diagram of Urea Production From NH3 and CO2himanshuchawla654No ratings yet
- AP Biology Practice Exam: Section I: Multiple-Choice QuestionsDocument83 pagesAP Biology Practice Exam: Section I: Multiple-Choice QuestionsKamilla DzhanzakovaNo ratings yet
- Manage Your Personal Energy PDFDocument46 pagesManage Your Personal Energy PDFezhil manickamNo ratings yet
- NCERT Class 3 English Baloon ManDocument10 pagesNCERT Class 3 English Baloon ManAnonymous 9WyPyismNo ratings yet
- Chemical Equilibrium and Le Chatelier's Principle: Chemistry 1Document17 pagesChemical Equilibrium and Le Chatelier's Principle: Chemistry 1azamatNo ratings yet
- Microscopy and StainingDocument7 pagesMicroscopy and StainingDenmark ManlusocNo ratings yet
- Human Genetics Concepts and Applications 11Th Edition Ricki Lewis Test Bank Full Chapter PDFDocument36 pagesHuman Genetics Concepts and Applications 11Th Edition Ricki Lewis Test Bank Full Chapter PDFdavid.cordero230100% (14)
- 6.1 MicrobiologyDocument32 pages6.1 MicrobiologyYsabelle BautistaNo ratings yet
- Prelim QuizDocument3 pagesPrelim QuizXyriel MacoyNo ratings yet
- Approved LaboratoriesDocument22 pagesApproved LaboratoriesPaul CNo ratings yet