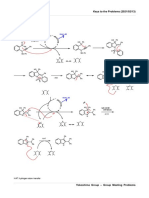
Professional Documents
Culture Documents
anti cancer drug
Uploaded by
Umashankar SharmaCopyright
Available Formats
Share this document
Did you find this document useful?
Is this content inappropriate?
Report this DocumentCopyright:
Available Formats
anti cancer drug
Uploaded by
Umashankar SharmaCopyright:
Available Formats
ANTI CANCER DRUGS
1. INTRODUCTION
Cancer is the second most leading cause for human mortality after cardiovascular disease.
According to the recent report of WHO, new cases have risen to 18.1 million in 2018.
Cancer is an uncontrolled growth of malignant cells that arise in any part of the body caused
by abnormal growth in normal cells. The symptoms of cancer are not general or specific and
mainly characterized by the type of cancer such as liver, lung, stomach, breast and colon
cancer are most common. Cancer is the most formidable affliction charac- terized by a
change in the mechanism that affects cell proliferation and differentiation resulted in solid
tumors including sarcomas, lymphomas and carcinomas [1]. One of the most fatal forms is
Leukaemia, which generally occurs through blood [2]. The main therapeutic treatment
includes chemotherapy, radiotherapy and surgery available for this most leading cause of
death worldwide. Chemotherapy associated with the use of low molecular weight drugs to
block or destroy cancer cells. A small molecular target can penetrate cancer cells and the
large molecule can attack as a whole to cancer cells, thus weaken them or can destroy their
enzymes involved in a mechanism [3]. Initial treatments were involving only surgical
methods. Chemotherapy became an attractive treatment since the 1940s with the development
of drug based upon nitrogen-mustard, a powerful class of alkylating agents and another
similar class in- cludes antimetabolites. The nitrogen-mustard analogues show activ- ity to
destroy cancer cells by attacking DNA and reduce its ability to replicate, while
antimetabolites arrest the S-phase of cell division responsible for the synthesis of DNA.
These findings lead towards the discovery of new potent chemotherapeutics [4]. The major
side effects associated with chemotherapeutic include hair loss, nausea, bone marrow
suppression, etc. because they have harmful effects on healthy cells along with cancer cells
[5]. Furthermore, the chemo- therapy drug treatment associated with the selectivity of
conven- tional chemotherapeutic agents and their acquirement by cancer cells for multiple
drug resistance is still a major limitation [6]. In an effort to combat chemotherapy, several
targeted therapies include modulator, apoptosis inducers, immunotherapies, angiogenesis
inhibitors, hormone therapy, Interleukin-6 antibodies therapy, toxin delivery molecule and
gene therapy, which have been used in can- cer treatment [7-10]. Several experimental
models for the study of cancer are available, which include cancer cell lines, xenografts,
tumor primary cell culture, paraffin-embedded samples and geneti- cally engineered mice
[11]. The cancer cell line model is considered to be an ideal model for the study of cancer due
to several advan- tages, including easy handling, instant accessibility, the similarity with an
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 1
ANTI CANCER DRUGS
initial tumor, highly homogeneous and reliability of results, high variation in availability
[12].
Heterocyclic compounds are an important class of organic chemistry as they are an integral
part of many drugs, natural prod- ucts and chemicals used in our daily life. Heterocyclic
compounds are composed of at least one heteroatom in a cyclic structure. Oxy- gen, nitrogen
and sulphur are the most frequently used hetero atoms.
O
O O O
S S
F3C S
NH2 NH2 N
S
N N O
O
O
Ethoxazolamide
Riluzole Probenazole
Cl
S H2N
NH
N N S
N
H
N O
Pramipexole
Ziprasidone
Fig. (1). Some marketed drugs bearing fused thiazole scaffolds.
Cl
N N HN
S
N N N S
O
H
N
HO
Levamisole Dasatinib
O
F S N
O
N
N
O
PMX610 YM-201627
Fig. (2). Anticancer drugs containing fused thiazole scaffolds.
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 2
ANTI CANCER DRUGS
Heterocyclic compound exhibits broad spectrum of pharma- cological activities such as
antitumor [14], antifungal [15], anti- inflammatory [16], anticonvulsant [17], and antibacterial
[18] activ- ity. In recent decades, efforts were made to design and synthesize less toxic and
more potent anticancer drugs with improved effi- ciency. Worldwide chemists reported
different anticancer agents with different heterocyclic scaffolds like benzimidazoles and
oxadi- azole [19], thiazoles and selenazole [20], pyrazoles and pyrimidine
[21] and imidazole [22]. Among heterocyclic compounds, thiazoles gained the attention of
medicinal chemists because of their broad spectrum of biological activities. Thiazoles are
five-membered heterocyclic compounds which contain Sulphur and Nitrogen at position-1
and position -3, respectively. Its chemical formula is C3H3NS. Thiazole is a clear yellow
colour liquid. Its boiling point is 116-118°C and specific gravity is 1.2. It is sparingly soluble
in water and soluble in alcohol and ether [23]. Naturally, thiazole has originated from
thiamine (vitamin B1), a water-soluble vitamin. Thiazole are an integral part of many potent
biological active drugs, including Isavuconazole (Antifungal) [24], Ritonavir (Anti-HIV)
[25], Nizatidine (Anti-Ulcer) [26]. Several anticancer agents bear- ing thiazole moiety also
reported in the literature, including Sulfa- thiazole, Bleomycin, Dasatinib, Epothilones,
Dabrafenib and Thia- zofurine [27]. Fused thiazole is a significant and extensively used
scaffold in drug designing and the development of novel therapeutic agents. Fused thiazole
scaffolds are considered to be a promising group of anticancer agents. Aryl group plays a
significant role in anticancer activity, but substitution at the different position might be
responsible for a better activity such as –Cl group at 3rd position of benzothiazole, phenyl and
some bulkier aryl group at imidazole thiazole ring, methoxy group at thiopyrano [2,3-d]
thiazole ring, napthyl substituent at phenothiazine ring included in this re- view. Some of the
marketed drugs bearing fused thiazole scaffolds are listed in Fig. (1), like Riluzole
(anticonvulsant), Ethoxazolamide (carbonic anhydrase inhibitor), Probenazole (herbicide),
Ziprasi- done (anti-psychotic), Pramipexole (parkinson’s inhibitor). Few anticancer drugs
containing fused thiazole scaffolds as marketed drugs and used in clinical trials are illustrated
in Figs. (2 and 3). Phortress was used in a xenograft model in phase 1 [28], voreloxin was
used in phase II for platinum-resistant ovarian cancer and acute myeloid leukemia [29],
Dasatinib was used in chronic myelogenous leukemia treatment, SNS-032 was used for
cyclin-dependent kinases and AC 220 as FLT3 inhibitor in phase III in clinical trials [30].
Benzo thiazole derivative 2-(4-amino-3-methylphenyl)-5- fluorobenzthiazole acting through
a novel mechanism, has also find importance in the clinical trial phase I in the UK [31].
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 3
ANTI CANCER DRUGS
2. SYNTHETIC STRATEGIES
Due to immense pharmacological importance, different synthetic roots have been
followed for the preparation of fused thiazoles using different starting substrate and
diverse reaction conditions. Various protocols for the synthesis of thiazole fused with six-
membered rings are summarized in Scheme 1.
In synthetic route 1, sulphanilamide 1 treated with chloroacetyl chloride 2 and
ammonium thiocyanate afforded substituted benzene sulfonamide, which further cyclized
after treatment with arylidine malanonitrile 3 and resulted in the formation of target
compound thiazolopyranopyrimidine derivative [32]. In route 2, a multistep strat- egy
was developed using an equal amount of o-aminothiophenol 4 and 2-amino-4-fluoro-
benzoic acid 5 refluxed with Polyphosphoric Acid (PPA) for 4h. The reaction mixture
after recrystallization was again refluxed with diethanolamine followed by thionyl
chloride to afford 2-[2-(N, N-dichloroethylamino)-4-fluoro phenyl]-benzothiozole as the
final product [33]. In another route 3, thiazolidinone 6 was treated with 1,4-
napthoquinone 7 via hetero-Diels-Alder (4+2)reaction to afford a series of novel
substituted 3,11-dihydro-2H-benzo [6, 7] thiochromeno [2,3-d][1,3]thiazole-2,5,10-
triones [34]. In synthetic route 4, the desired product obtained by the heating of naptha
[1,2- d]thiazol-2-amine 8 with substituted aromatic aldehyde 9 using acetic acid as a
reaction medium. After refluxing the content, the reaction mixture was poured onto ice to
obtain a solid product, which further recrystallized with ethanol to afford the desired
prod- uct [35]. In synthetic route 5, a multistep procedure is reported treating o-amino
thiophenol 10 and diethyl oxalate 11 under reflux for 8h. The solid product obtained after
recrystallization, reacting with lithium hydroxide monohydrate in THF and water at
ambient temperature. The solid was added to DMF, phenyl alanine deriva- tive 12,
HATU (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo [4,5-b]pyridinium-3-oxo
hexafluorophosphate) and DIPEA (N,N- diisopropylethylamine) at 0°C. Then the whole
mixture was stirred for 10h at room temperature. The desired product was obtained from
the organic layer after washing with brine solution and dilute HCl [36]. In route 6, 6-
subsituted aniline-2-thiouracils 13 were alkylated at sulphur, and in the presence of
K2CO3 afforded S- alkylated 2-thiouracils, an intermediate. The target compound was
obtained by intramolecular cyclization using conc. sulphuric acid at 20°C [37]. In route 7,
substituted aniline 14 was treated with am- monium thiocyanate and bromine in
chloroform. The intermediate is formed by the nucleophilic attack of amine at the
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 4
ANTI CANCER DRUGS
electrophilic carbon of thiocyanate ion. The final target compounds were af- forded by
the cyclization in the presence of bromine and chloro- form [38]. In synthetic route 8, the
Mannich reaction mechanism was followed in which 2-iminothiazolidine-4-one 15
reacted with formalin and aniline in a molar ratio 1:2:1 yielded the desired prod- uct [39].
In route 9, the synthesis of the target compound was re- ported in two steps. In this
strategy, 2-amino-4-(substituted phenyl) thiazole 16 was refluxed with cyanoketone 17 in
DMF in the pres- ence of Triethylamine (TEA). The intermediate thus obtained was
refluxed with hydrazine hydrate in ethanol for 3h to afford fused thiazole compound as
the desired product [40]. In route 10, 3,4- dihydropyrimidine-2-thione 18 was treated
with α-haloester 19 under reflux in ethanol to afford the product with excellent yield
[41]. The synthetic route 11 was a two-step protocol consisting of the treatment of 2-
aminothiophenol 20 with p-anthranilic acid 21 in the presence of molecular iodine as a
catalyst to afford the cyclized intermediate which on further heating with substituted
sulphonyl chloride 22 and acetic anhydride afforded the desired product [42].
Similarly, various strategies for the synthesis of thiazole fused with five-membered
rings are summarized in Scheme 2. In syn-thetic route 12, PBTz was prepared via
Buchwald Hartwig coupling between a branched alkyl amine and silylated bromide in a
sealed tube at 170°C yielded intermediate 2, 2′-dibromo-5, 5′-bithiazole 23. Then
deprotection was done using TBAF, which then exposed to NBS in DMF directly to be
bromination to yield PBTz
S
O N
NH
H O N
S N H
N N
S N
N O HO N
O O
SNS-032 (BMS-387032)
Voreloxin
CH3
R S O S
N
NH H
(CH2)4 NH2 O N
N N
O NH2.2HCl
O N O
Phortres
AC 220
s
N N
H H
Fig. (3). Anticancer drugs containing fused thiazole scaffolds currently in clinical trials.
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 5
ANTI CANCER DRUGS
Route1 COOH Route 1
Cl O
1 NH2 SO2C
Route NC
10 + 2
O + + C
Cl
Ar SH C R1
O R H
O NH + 20 R
21 22
H 2N O +NH2
R1 O O R2
N S S
H 3 R3
18 19
Route9
NH2 HOOC
O
SM +
EtO SH Cl F
SM
CN 4 5
17
+
R1
R4 Route 3 O
NH 2 H
S S
N
N
R3 O
N +
1,4-napthoquinone
S S
16 AcOH,hydroquinon
ecatalyst,1h
R2 Ar
6 O
7
H S NH2 Route4
H2 N
HN O
N CHO
N
S
15 +
Route
9 R
8 8
O
R1 R
NH O OEt
R2 NH2 H 2N O
NH2 +
R1 N S
H O
OE H2N HN R1
14
13 + 11
10 12
Route 7 Route 5
Scheme 2. Synthetic strategies of thiazoles fused with five-membered rings
In synthetic route 13, 3-substituted pyrazole-4-carbaldehyde 24 was
oxidized with an alkali solution of potassium permanganate to yield the corresponding acid.
The acid thus obtained was condensed with 3-substituted-4-amino-5-mercapto-1, 2, 4-
triazoles 25 in the pres- ence of phosphorus oxychloride to afford the desired product [44]. In
route 14, the target compound was obtained by the condensation of 2-amino-5-substituted-
1,3,4-thiadiazoles 26 with α-bromoketone. The synthesis was done under reflux for 8-10h
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 6
ANTI CANCER DRUGS
followed by neu- tralization with cold aqueous sodium carbonate solution, which afforded the
product with good yield. The alternate route was fol- lowed for the same reaction using
microwave irradiation in DMF for 4-7min.
Leukemia
Tryosin kinase inhibitors Colon cancer inhibitors
CEM, HL60, U937
inhibitors
Tubulin inhibitors
CNS SNB -75 inhibitors
R1
R2
CDK1 inhibitors
N
Melanoma cancer
cell inhibitors
Fused Thiazole
Fox M1 inhibitors
Cell cycle arrest inhibitors
Angeogenesis inhibitors
Breast cancer
Ovarian cancer cell inhibitors
cell inhibitors
Dual Src/ AbI kinase inhibitors
Fig. (4). Mechanism of action of fused thiazoles as anticancer agents
The substrate used could be prepared by the reaction of aryl substituted acetic acid 27 and
thiosemicarbazide 28 in sul- phuric acid on heating up to 70°C [45]. In route 15, the
preparation of hydrate hydrazones was outlined. In this procedure, ethyl 6-(4-
bromophenyl)imidazo[2,1-b]thiazole-3-acetate 29 was refluxed with hydrate hydrazine to
give intermediate[6-(4-bromophenyl)imidazo [2,1-b]thiazol-3-yl]acetic acid hydrazide which
then condensed with a suitable aromatic aldehyde to afford the target compound [46]. In
route 16, ethyl-2-aminothiazole-4-acetate 30 was reacted with 4-chloro-2-
bromoacetophenone 31 and acetone in the presence of ethanol to obtain ethyl-6-(4-
chlorophrnyl imidazo [2,1-b]thiazol- 3-acetate hydrobromide. It was a key intermediate and
can be fur- ther used for the preparation of various derivatives [47]. In route 17, dithioxamide
32 in nitrobenzene was stirred with aldehyde 33 at 130°C for 24h under inert nitrogen
atmosphere to afford 2,5- disubstituted thiazole[5,4-d] [48]. In route 18, the target compound
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 7
ANTI CANCER DRUGS
was obtained by the 1,3 dipolar cycloaddition reaction of thia- zolium derivative 34 with
electron deficient alkene 35. The reaction was catalyzed by a base and terakispyridinecobalt
(ll) dichromate [PyCo(HCrO4)2 [49]. In route 19, various 2-aminothiazoles 36 were cyclized
in the presence of ethyl bromopyruvate. The intermediate thus obtained afforded the target
compound imidazo [2,1- b]thiazole-6-carboxylic acid by chlorination, followed by hydroly-
sis with lithium hydroxide [50]. In approach 20, the starting mate- rial 3-[1-(4-(2-
methylpropyl) phenyl) ethyl]-1,2,4triazole-5-thione 37 was prepared by Ibuprofen. It was
cyclized with chloroacetic acid 38 and suitable aldehyde 39 in the presence of acetic acid,
acetic anhydride and sodium acetate [51].
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 8
ANTI CANCER DRUGS
3 . FUSED THIAZOLES AND THEIR ANTICANCER PROPERTIES
Fused thiazoles are one of the key scaffolds among heterocyclic compounds and had
drawn the attention of medicinal chemists due to their potent chemotherapeutic activities.
Several members are reported in the literature as effective agents in chemotherapy. A
different mechanism of action is followed by fused thiazoles for their anticancer activity,
which includes inhibition of certain en- zymes like Src and AbI kinase and cytochrome.
Similarly, fused thiazole targets different cancer cells such as MCF-7 (human breast
cancer), HEPG2 (human hepatocellular liver-carcinoma), CNS (central nervous
systemcancer ), SNB-75, Renal UO-31, leukaemia, colon, melanoma (skin cancer),
ovarian, prostate, breast cancer, etc., as shown in Fig. (4). Preliminary screening of the
target com- pound revealed the significant inhibitory activity against different cancer cell
lines. In this review, we will elaborate various types of fused thiazole as an anticancer
agent.
Benzothiazole
Benzothiazole (BT) has become a privileged scaffold over the last two decades
because of its wonderful pharmacological profile, including anticancer activity. Further
substitution and other rings fused with benzothiazole increased its activity. In 2019,
Shokrollahi et al. designed Schiff based fused thiazole from 4,5,6,7 tetrahydro-
benzo[d]thiazole-2,6-diamine and aldehydes, as depicted in Fig. (5). Furthermore,
compounds 40-43 were evaluated for their anticancer potential against human cancer
cells MCF-7 and HepG2 using MTT based assay and compound 40 was found to be most
potent against both MCF-7 and HepG2 cancer cells after an incubation period of 48hrs,
as shown by IC50 values in Table 1. In addition, the target compound was able to bind
with Human Serum Albumin, con- cluded from the computational docking method and
experimental fluorescence quenching and circular dichroism method and thus possessed
one binding site [52].
In 2019, Reddy et al. prepared a series of pyrazole linked ben- zothiazole compound
44-45, as shown in Fig. (6). All the tested compounds have shown potential inhibition
against VEFGR-2 pro- tein and four other cancer cell lines, namely colon (HT29),
prostate (PC3), lung (A549), kidney cell (Hek-293T), brain cell (U87MG). Compound
44-45 exhibited VEFGR-2 inhibition with IC50 value 97nM and 109nM, respectively,
compared with standard reference compound Axitinib with an IC50 value of 39nM.
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 9
ANTI CANCER DRUGS
Further docking study reveals the interaction of the active site of VEFGR-2 protein with
compounds 44 and 45, which can easily fit into binding pock- ets of VEFGR-2 protein.
Compound 44 also acted as a potent in- hibitor of U87MG and PC-3 multicellular
spheroids and increased G0/G1 population, arrest cell cycle and induced apoptosis through
mitochondrial function [53].
In 2019, Mishra et al. reported a Schiff base linked with ben- zothiazole 46a and
explored its MCF-7 inhibitory and DNA cleav- age activity with a standard reference
compound tamoxifen. A fur- ther calorimetric study using MTT based assay reveals that
com- pound 46a, as shown in Fig. (7), exhibited approximately 86% in- hibition against
MCF-7 breast cancer cells with IC50 value 150µg/ml and against normal cells IC50 value
973µg/ml. In addi- tion, docking studies reveal the interaction of compound 46 with
EGFR tyrosine kinase domain and in silico pharmacokinetic study of compound 46 done
with Swiss ADME server explored inhibition against certain cytochrome enzymes
including CYP2C19, P-gp substrate, CYP2D6 and CYPC9, which proves it a promising
lead for the development of anticancer drugs [54].
In 2019, Hassan and his coworkers synthesized benzothiazole derivatives by multistep
chemical reaction starting from 2- (Benzo[d]thiazol-2-yl) acetonitrile, as depicted in Fig.
(7). Further- more, among the series, thirteen compounds were selected for in vitro
anticancer screening at NCI institute against a panel of sixty cancer cell lines.
nd
Compound 46 b-c substituted benzothiazole 2 position with 3-amino-5-
phenylaminopyrazole and 3-amino-5- phenylaminothiophen-2-yl-(4-chlorophenyl)
methanone group showed significant inhibition with an IC50 value ranging from 0.683 to
4.66μ/mL against a full panel of cancer cell lines [55].
In 2018, Zehra et al. reported the synthesis of benzothiazole Schiff bases ligands
complexes by the refluxing of different ben- zothiazoles with o-vanillin 47-48 followed
by complexation with Cu(II) and Zn(II), as shown in Fig. (8) and explored their
anticancer activity against different human cancer cells named HepG2, HeLa, A498,
MCF-7 and MIA-PA-CA-2. Further SAR and docking study reveals that chlorine
substituent is responsible for the inhibition and cytotoxic activity as it increases the
hydrophobic and lyophilic character of the target compound. Moreover, the complex with
copper metal 47 shows excellent inhibition against HeLa cancer cell with an IC50 value of
4.8 μg/ml, while the complex with zinc metal 48 was almost inactive as proved by
cytotoxic profile.
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 10
ANTI CANCER DRUGS
OH
R1 OH
N
N
N
N
S R2
N R2 S
N
40 HO
HO
41-43
R1
Fig. (5). Schiff base derived benzothiazole.
Compound No. R1 R2 MCF-7 HepG2
40 - - 3.01±2.71 1.29±3.07
41 H H 15.39±0.86 51.19±4.86
42 H Br 11.15±0.45 43.14±4.23
43 OMe H 14.26±0.41 43.99±4.21
Table 1. IC50 value (in µM) of Schiff base derived benzothiazole.
when R1= F and R2= Cl compound exhibit significant
VEGFR-2 inhibition
R1
N
HN
N
S R2
N
O
44-45
Fig. (6). Pyrazolo linked benzothiazole derivative as VEFGR-2 inhibitors.
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 11
ANTI CANCER DRUGS
H 2N
(Et)2N OH S
N N NH
N NH S N
N
S
HN O
S
OH
NH2
46a 46b 46c
Cl
Fig. (7). Benzothiazole derivatives as cancer cell line inhibitors.
Thiazole Fused with the Imidazole Ring
Imidazoles are well-known privileged scaffolds as a large part of these molecules have
numerous applications in the medicinal field. A plethora of compounds are reported in
the literature as anti- bacterial, antifungal, anthelmintic and antitumor activity [57]. Le-
vamisole is a potent anticancer drug reported in the literature bear- ing imidazole-thiazole
moiety.
In 2019, Nagireddy et al. reported the synthesis of Noscapine coupled derivatives 49(a-f),
as shown in Fig. (9) with imidazo[2,1- b]triazole from naturally occuring alkaloid
Noscapine and explored their anticancer activity against different cancer cells named
MIA- PaCa-2 (pancreatic), SK-N-SH (neuroblastoma), DU-145 (pros- tate), and MCF-
7(breast), using the SRB assay. It was evaluated that o-imidazothiazolyl noscapinoids
were more potent than N- imidazothiazolyl noscapinoids towards the inhibitory activity.
Flow cytometry study further showed that compounds 49b-f arrest cell cycle in the G2/M
phase. All the tested compounds caused signifi- cant degradation in caspase -3 and PARP
concentration, which induced cell apoptosis in MIAPaCa-2 cancer cells. Molecular mod-
elling illustrates the binding interaction of tubulin protein with the target compound and
revealed that substitution of the methoxy group at the 6th position of the phthalide ring
responsible for anti- cancer activity [58].
In 2016, Karaman and coworkers prepared the imidazole- thiazole compound bearing
hydrazone moiety by the reaction of ethyl-2-amionothiazole-4-acetate with 4-chloro-2’-
bromoacetophenone in acetone, the intermediate formed was treated with hydrazine
hydrate followed by aromatic aldehyde to afford the target com- pound arylidene
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 12
ANTI CANCER DRUGS
hydrazide. Furthermore, the antiproliferative activity was evaluated against OVCAR-3,
HCT-15, CCRF-CEM,SR, Uo-31 and CAKI-1 cancer cell lines. The most significant
activity was shown by compound 49g, as illustrated in Fig. (10). Compound 49g showed
potent growth inhibition on ovarian cancer cell includes OVCAR-3 and OVCAR-4 than
sorafenib, while having a poor cy- totoxic effect on all melanoma cancer cells than
sorafenib.
Cl group increase inhibition againstHeLa cancer cell than H group
Cl
H3CO OH N
47-48
Fig. (8). Schiff base bearing benzothiazole.
In 2014, Kamal et al. synthesized chalcone hybrid bearing imi- dazo [2,1-b]thiazole moiety
using benzoin as a starting material. An anticancer evaluation was done using different cancer
cells against doxorubicin as a standard drug, compound 50 was found to show the most
promising inhibition against lung cancer cell (A549). Thus, A549 cancer cell was selected to
study the cell cycle inhibi- tion and tubulin polymerisation inhibition. Furthermore, confocal
microscopy of immunohistochemistry studies of tubulin illustrates the inhibition of A549
cells as a perfect order network was found in control cells than the target compound and
standard drug Nocoda- zole used. Compound 50 was able to bind with tubulin protein ef-
fectively with IC50 value 1.43µM, while the reference compound Nocodazole showed IC50
value 1.23µM, as illustrated in Fig. (11). In addition, compound 50 was also able to bind cell
cycle progres- sion in the G2/M phase via inducing centrosome formation and abnormal
spindle structure, which confirmed its anti-proliferative activity [59].
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 13
ANTI CANCER DRUGS
In 2014, Koppireddi and his team reported 3, 6-diphenylimidazo [2,1-b]thiazole derivatives
(51-52), as shown in Fig. (12). Com- pound 51 was evaluated for anticancer activity against
HeLa cancer cells. The trifluorophenyl group is responsible for promising cell proliferative
activity of compound 51 exhibiting 6.5±0.56µM IC50 value with reference to standard drug
doxorubicin with IC50 value 6.0±0.7µM. Furthermore, compound 51 was able to hinder the
cell cycle in the G0/G1 phase and induced apoptosis. Cell apoptosis was also confirmed by
caspase -3 ad caspase -8 activities of the cell [60]. Similarly, compound 52 bearing pyrazole
moiety was pre- pared by the reaction of 6-hydrazinylimidazo[2,1-b]thiazoles with diketones
and exhibited a comparable response like mTOR inhibi- tor Rapamycin against CNS SNB-75
and Renal UO-31 cancer cell lines. Compare analysis was done to explore their anticancer
drug like cytotoxicity, Lipinski’s rule of five, calculated by Osiris and Canvas programs, was
an important tool used to illustrate their drug like ADME properties and bioavailability [61].
In 2015, Patel et al. synthesized a novel and potent ALK5 in- hibitor 53 containing
thiadiazole fused with the imidazole ring, as illustrated in Fig. (13). Further docking study
reveals that the sul- phur atom of the thiadiazole ring forms strong interactions with His-283
in the hinge region of kinase, the binding site of the target. In addition, SAR study reveals
that electron-withdrawing group like fluorine on the phenyl ring contributes to ALK5
inhibition [62]. Other thiadiazole and imidazole hybrid molecules 54-56 containing m-(α-
bromoacryloylamido) phenyl moiety linked at the 6th position of the imidazole-thiadiazole
ring were reported by Romangoli and his coworkers, as depicted in Fig. (14). The reported
compounds 54-56 are found to be most effective to bring about apoptosis among the series
prepared with IC50 value comparable or lower than standard reference compound Melphalan,
as summarized in Table 2. The antiproliferative activity was associated with the release of
cyto- chrome-c and cleavage of caspases [63].
In 2014, Gali et al. reported the synthesis of coumarinylimi- dazo[2,1-b]thiazole derivatives
57-60 via Vilsmeier-Haack and Knoevenagel condensation reaction and explored in vitro
anticancer activity against HepG2, HeLa, MCF-7 and NCl-H460 cancer cell lines.
Furthermore, compound 60 showed a significant broad- spectrum activity towards all cancer
cell lines, as shown in Fig. (15) and detailed activity in terms of IC50 value against various
cell lines using doxorubicin as a standard reference is illustrated in Table 3 [64].
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 14
ANTI CANCER DRUGS
O
O
F OMe
N Cl
N
O N O N
N H
S O
N S
O
O
H O
MeO H
OMe
H
O OMe Br
N
O O
N N
OMe 49b H
O OMe S
O
O
49a H
O OMe 49c
O
OMe O
O
OMe
S
N OMe Br
N O N
H
N
S N O N CH3
O H
O
H S
O
OMe O
O H
O 49d
O OMe 49f
O
Fig. (9). Noscapine coupled derivatives with imidazo [2,1-b]thiazole
Cl
N NH N
HO
N
S
H
49
shows higher cytotoxic effecton ovarian cancer cell
line compared to Sorafenib
Fig. (10). Arylidenehydrazide compounds as ovarian cancer cell inhibitors.
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 15
ANTI CANCER DRUGS
MeO
N
N
MeO F
O
Group responsible to inhibit
N
50
tubulin protein by interacting
colchicine binding site.
Fig. (11). Significant tubulin inhibitor.
R when R=H, R1= O
N N
H
R1
N Show significant anticancer activity towards
N CNS SNB-75and Renal UO-31cancer cell lines
51-52
When R= CF3,R1= C6H5 shows comparable
anticancer activity with doxorubicin
against HeLa cells
Fig. (12). Different anticancer targets containing imidazo [2,1-b]thiazole
Compound No. R HL-60 U937 U937/Bcl-2 SK-MEL-1
54 Thien-2-yl 0.38±0.05 0.57±0.04 0.27±0.16 0.69±0.29
55 m,p-(OCH3)2- 0.21±0.09 0.27±0.11 0.21±0.13 0.77±0.13
C6H3
56 p-F-C6H3 0.53±0.16 0.56±0.20 0.37±0.16 1.10±0.22
Table 2. IC50 value (in µM) of the Thiadiazole-imidazole tethered compound.
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 16
ANTI CANCER DRUGS
when R= p-OC2H5-C6H4 and p-OCH3-C6H4 shows
similar effect on all cancer cells
R N
S H
N
N Br
54-56
Fig. (14). Thiadiazole-imidazole tethered compound as a potent ALK5 inhibitor.
HN
O O R
N
N
57-60
Fig. (15). Coumarinylimidazo[2,1-b]thiazole derivatives.
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 17
ANTI CANCER DRUGS
Thiazole Fused with the Pyrimidines Ring
Pyrimidine derivatives are a significant type of heterocyclic compound, which exhibited a
number of pharmacological profiles such as antifungal [65], antitumor [66], antibacterial
[67], and anti- inflammatory [68]. Pyridine fused with the thiazole ring with dif- ferent
substitution showed remarkable biological properties. Thia- zolo [3,2-a]pyrimidines have
gained importance in medicinal chem-istry as it is a basic building block of nucleotide. In
addition, ritan- serin, a serotonin receptor antagonist and setoperone, is a ligand of 5-HT2A
receptor containing thiazolopyrimidine scaffolds [69].
In 2017, Hassan et al. reported the synthesis of thiazole [4,5- d]pyrimidines and evaluated
their antitumor activity using a stan- dard reference compound doxorubicin. The results
concluded that compound 61 was found to be most active towards breast, prostate, renal,
ovarian, non-small cell lung, colon, CNS and leukemia can- cer cells lines, as depicted in Fig.
(16). Furthermore, the SAR correlation of the compound reveals that the presence of amine
and methoxy group at phenyl ring increases the activity of analogs than unsubstituted
congener while the presence of the electron- withdrawing group like chlorine decreases the
activity ten times than other analogs. In addition, molecular modelling reveals that
compound substituted with the amine and trimethoxy group at the phenyl ring showed
binding interaction with DNA bases like ade- nine, cytosine and thiamine and the study was
compared using a standard reference compound distamycin [70].
In 2017, Yousif et al. synthesized new thiazolopyrimidine de- rivative and explored their
anticancer activity against HepG-2, HCT-116 and PC-3 cancer cell lines. Compound 62a
with IC50 value 66.5±3.6µg/mL was found to be more potent and showed higher cytotoxic
activity by dose-dependent behaviour than stan- dard compound doxorubicin with IC 50 value
75.24±4.1µg/mL against PC-3 cancer cells, while for other two cancer cells exhibited weak
to moderate cytotoxicity, as illustrated in Fig. (17). Further- more, substitution at pyrimidine
nucleus demonstrates a significant effect on the cytotoxic activity of the target compound
[71]. Basiony and his coworkers also reported the synthesis of thia- zolopyrimidine
substituted with thienyl- or chlorophenyl- deriva- tives by incorporating sugar hydrazone
moiety. Derivatives 62(b-e) prepared and screened for their anticancer activity against
different cancer cells include Caco-2, MDA-MB-231, HCT-116 and MCF-7 cancer cells, as
depicted in Fig. (18). Compound 62 (b-c, e) pos- sessed the lowest IC50 value 9.63, 4.79,
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 18
ANTI CANCER DRUGS
16.82µg/mL, respectively and exhibited higher cytotoxicity with a regular decrease in cell
proliferation than others in a dose-dependent manner against Caco-
2 cells. Compound 62(d) also showed better inhibition against MDA-MB-231 cells with the
lowest IC50 value of 23.35µg/mL. It was further concluded that sugar hydrazone derivative
62(b,c) with the hydroxyl and xylosyl group was more potent than 62(d,e) with an acetyl
group. The cytotoxicity was also elaborated with thia- zolopyrimidine substituted with
thienyl- or chlorophenyl- deriva- tives without sugar moiety, which were proved to be more
cytotoxic against HCT-116 cancer cells [72].
In 2011, Abu-Hashem and coworkers synthesized diaryldioxa- zolo-pyrrolothiazolo-
pyrimidine derivatives and evaluated their in vitro cytotoxic effect. Furthermore, the SAR
study concluded that compound diaryl-dioxo-pyrrolo-thiazolo-pyrimidines deriva- tives 63-
65 are more potent cytotoxic candidates than pyrimidine derivatives, as shown in Fig. (19).
The cytotoxicity was compared with a standard reference compound 5-flououracil, as
summarized in Table 4 [73].
Compound No. R X MCF-7 HepG-2 HeLa NCL- H460
57 4-BrC6H4 O 21.00±0.8 30.00±0.5 7.13±0.4 43.25±0.7
58 4-ClC6H4 O 39.45±0.3 27.01±0.7 45±0.8 26.29±0.1
59 4-BrC6H4 S 25.99±1.0 42.047±0.5 14.21±0.6 136.04±0.6
60 4-ClC6H4 S 16.99±0.7 13.92±0.2 5.18±0.1 32.37±0.9
Standard Doxorubicin - 3.00±0.3 1.10±0.06 1.33±0.11 0.83±0.03
Table 3. IC50 value (in µM)of Coumarinylimidazo[2,1-b]thiazole derivatives.
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 19
ANTI CANCER DRUGS
Thiazole Fused with Quinazoline
Thiazoles condensed with quinazoline are a vital class of het- erocyclic compounds and
possess a wide spectrum of biological properties. Al-omary et al. reported the preparation of
thiazolo [2,3- b]quinazoline derivatives 66-75, as shown in Fig. (20) and evalu- ated their
anticancer properties. Compound 68 is the most active compound and exhibited broad-
spectrum activity with a GI50 value
2.5 lower than standard compound 5-flourouracil, but the introduc- tion of the methyl group
to 68 leads to an inactive compound 67. The unsubstituted compound 66 and methoxy
substituent 75 are mostly inactive compounds proved by structure-activity relation- ship. The
position of the methyl group also serves a useful parame- ter. Compounds 69 and 72 with
methyl substituent showed moder- ate anticancer activity, while compound 73 becomes
inactive.
In 2014, Gali et al. prepared a series of indolyl methylene benzo[h]thiazole [2,3-
b]quinazolinones derivatives from benzo[h] quinazoline-thione derivative reacted with 2-
chloro-N-phenylacet- amide and indole-3-carbaldehyde via Knoevenagel condensation
reaction. In addition, all synthesized compounds were evaluated against MCF-7 and HepG2
cancer cell lines using thestandard drug doxorubicin and compounds 76-78 have shown
significant activity, as shown in Fig. (21) and their detailed IC50 value is summarized in Table
5. Compound 76 has proved an excellent inhibitor against MCF-7 cells and 77 against both
cells, including MCF-7 and HepG2 cancer cells. It was further concluded that 5-bromo indol-
3-ylmethylene substituent on the thiazole ring and 4-chlorophenyl on the pyrimidine ring are
responsible for significant inhibitory activity [75].
Compound No. R Dead cell %
63 Phenyl 50.10
64 p-chlorophenyl 65.14
65 p- methoxyphenyl 55.17
5-Fluoro uracil - 99.50
Table 4. Cytotoxic comparison of thiazolopyrimidines derivative
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 20
ANTI CANCER DRUGS
Thiazole Fused with the Phenothiazine Ring
Phenothiazines are important tricyclic heteroaromatic com- pounds and considered to be a
key scaffold due to their diverse pharmacological properties. In 2017, Brem and coworkers
synthe- sized thiazole hybrid compounds with phenothiazine by oxidative cyclisation and
evaluated their anticancer activity against HL-60 human promyelocytic and THP-1 human
monocytic leukaemia cancer cell lines. Furthermore, napthyl substituted compound 79
showed comparable antiproliferative activity than the standard drug cytarabine, while nitro
phenyl substituent decreases the activity, as shown in Fig. (22). The target compound induced
apoptosis by inhibiting mitochondrial DNA polymerase, caspase fragmentation and decreased
ATP formation.
Ar Ar
HO O N AcO O N
S n(AcOHC)
n(HOHC) S
N NH N N NH N
62(b-c)
62(d-e)
sugar moiety with n- b,c=D-
where Ar- xylotetritolyl; n=3
b,d=4-chlorophenylc,e d,e=penta-Oacetyl-galactopentitolyl;n=4
Fig. (18). Thiazolopyrimidine derivative incorporating sugar moiety.
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 21
ANTI CANCER DRUGS
H3CO
S
S
H3CO
H3CO N CH3
N
N N
N S
H3CO OCH 3
N
H3CO
CH3 OCH3
Inactive Inactive 67
Inactive H 3CO
66
75 OCH3 S
H 3CO
N
OCH 3 N
H3CO
H3CO
S
H3CO N OCH 3
N
S most active
OCH 3
H3CO
moderate active R4
N N
OCH 3
74
R1 R1 68
OCH 3
S S
H3CO R2 R2
N
CH 3
N R3 R3 N N
CH3
Inactve
73
moderate active 69
OCH 3 S
H3CO S
S
N
CH3 N N
N N
N
moderate active
72 Cl Cl
Cl Cl
70
71
OCH 3
Fig. (20). Structure of thiazolo [2,3-b]quinazoline derivative as anticancer agents
Thiazole Fused with Thiopyrano Moiety
Thiazole compound fused with thiopyrano is of great interest as it leads to discover new
anticancer agents. In 2015, Metwally et al. reported thiopyrano [2,3-d]thiazole derivatives
80-82 by the Knoevenagal reaction of substituted thioazolidinone with pyrazole-4-
carbaldehyde followed by cycloaddition reaction with acryloni- trile, ethylacrylate and N-
arylmaleimides. Further anticancer activ- ity was evaluated against MCF-7 and HepG2
cancer cell lines, as shown in Fig. (23). Docking study reveals that substitution by an
electron-withdrawing chlorine group decreases the activity towards both MCF-7 and HepG2
cancer cell lines by inducing similar mechanism to that of tubulin polymerisation inhibitors
while substi- tution with an electron-donating methoxy group decreases the sen- sitivity of
cancer cell lines, as summarized in Table 6.
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 22
ANTI CANCER DRUGS
R1
N
Br
S
N
N
H3CO
76-78
Fig. (21). Indolylmethylene derivative as an anticancer agent.
Compound No. R1 MCF-7 HepG2
76 Cl 2.59 11.55
77 OCH3 7.72 8.60
78 F 8.96 22.1
Doxorubicin 3.00 1.10
Table 5. IC50 value (in µM) of compound 76-78 against two different cancer cell
lines.
In 2014, lozyanski and coworkers synthesized a series of thiopyrano and thiazole hybrid
compounds with cinnamic acid de- rivative by hetero Diels Alder reaction and their
anticancer activity was evaluated against a panel of 60 cancer cell lines. Furthermore, the
SAR study reveals that methoxy and chloro substituent present
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 23
ANTI CANCER DRUGS
when R= 1-C10H7,
compound is most
active
N
S
N
S
when R= 4-NO2-C6H4 ,
compound is inactive
towards antiproliferative activity
79
Fig. (22). Structure of thiazole-phenothiazine derivative as an antiprolifera- tive agent.
when R1= OCH3, MCF-7 aremost sensitive than others
O Ph
H
S N
R1 N O
SN
HH N
O
Ph
80-82
Fig. (23). Thiopyrano fused with thiazole.
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 24
ANTI CANCER DRUGS
H
N O
HN R
when R = 4-Cl-C6H 4, R1=Me and Cl, showed
O
more cytotoxic effect
R1
83
Fig. (24). Thiopyrano [2,3-d]thiazoles derived from cinnamic acid amide
OH
MeO
N
S
H
84
Fig. (25). Thiopyrano [2,3-d]thiazole containing napthoquinone
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 25
ANTI CANCER DRUGS
Fig. (26). Thiazole- androstenones hybrid as anticancer agents.
H H
R
when R=
F
F F
H H
N N
85 86
87
Compound No. R1 MCF-7 HepG2
80 H 22.4 15.8
81 OCH3 12.3 21.4
82 Cl 35.9 31.3
Table 6. IC50 values (in µg/ml) of compound 80-82.
at the amide fragment increases the activity of compound 83, as shown in Fig. (24) [78]. In
addition, Atamanyuk et al. also reported thiopyrano [2,3-d]thiazoles 84 by hetero-Diels Alder
reaction con- taining napthoquinone fragments and explored their antineoplastic activity
against UACC-257- melanoma cancer cell lines as, illus- trated in Fig. (25). The mechanism
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 26
ANTI CANCER DRUGS
of action of the target compound was explored using COMPARE analysis and found the
similarity of 84 in cytotoxicity with some known anticancer agents including S- trityl-L-
cysteine, didemnin B and panсratistatin [79].
Thiazole-Steroidal Hybrid Derivative
Steroids are an important class of natural products that control various cellular functions
including cell proliferation, apoptosis, homeostasis and differentiation. A wide range of
steroids were extracted from plants and microorganisms and showed a broad spectrum of
biological activity. Heterocyclic attached or fused ster- oid compounds such as androstanes,
oleandrigenin and galeterone are known to exhibit diverse pharmacological profiles.
In 2018, Ali et al. prepared aminothiazole-androstenone deriva- tives by the reaction of 6β-
bromoandrostenedion with different substituted thiourea and further evaluated their
anticancer activity against a panel of 60 cancer cell lines. Compounds 85-86 display
inhibition against different cancer cells, including colon, CNS, SF- 295, SF-539, melanoma
and renal cancer cells at 50% inhibitory concentration value less than 2µM [80]. In 2018,
Okolo also re-ported the aminothiazole-androstenones derivatives by simple methodology, in
which 6β-bromo androstenedione reacted with thioamides in hexafluoroisopropanol as a
reaction medium. In addi- tion, among the reported derivatives, compound 87 proved to be
most potent against 31 cancer cell lines out of 60 cancer cell lines at 50% inhibitory
concentration value less than 2.58 µM, as shown in Fig. (26) [81].
In 2016, Cui et al. evaluated the antitumor activity of D-ring fused 1,2,3-thiadiazole
dehydroepiandrosterine (DHEA) derivatives against various tumor cells, including breast
cancer cells, colon carcinoma cells, prostate cell, T lymphocyte cells, promyelocytic, and
leukemia cancer cell lines using the SRB assay. The antitumor activity was analysed using
different models subcutaneous xenograft growth model, metastasis model and orthotopic
xenograft growth. In addition, among all the tested compounds, significant activity exhibited
by only compound 88, as shown in Fig. (27) against human breast cancer cells (T47D) and all
other compounds were inactive towards all cancer cell lines, as shown in Table 7.
Furthermore, the primary mechanism study of compound 88 dem- onstrates that it induces
apoptosis by dose-dependent manner and phosphorylation of EphB3 and EphA2, but the cell
cycle arrest mechanism was not explored [82].
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 27
ANTI CANCER DRUGS
S
N
NH
88
Fig. (27). DHEA derivative as an antitumor agent
Table 7. IC50 value of DHEA and its derivative.
Compound No. T47D HAF SI
88 0.058±0.016 21.1±5.06 364
DHEA 2.55±0.001 >50 ND
ADM 0.040±0.018 0.068±0.064 1.7
S S
OH OH
N N
HO Me Me
Me
S Me
S
OH
N
OH
N
Me
Me
91
92
Fig. (28). Pyrrolo [1,2-c]thiazoles derivatives as TN inhibitors
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 28
ANTI CANCER DRUGS
Pyrrolo-Thiazoles Hybrid Compound
Pyrrolo [1,2-c]thiazoles are an important aromatic scaffold used for designing new promising
anticancer drug candidates. In 2014, Santosh et al. reported 6,7-bis(hydroxymethyl)-1H, 3H-
pyrrolo[1,2- c]thiazole derivatives, significant inhibitors of Triple-Negative (TN) breast
cancer cells. Compounds 89-92 are promising inhibi- tors against TN (HCC1806), MCF7,
and HCC1954 breast cancer cell lines, as shown in Fig. (28). Furthermore, it was concluded
from the SAR study that the hydroxy phenyl group at C-3 of the thiazole ring proves it most
promising TN inhibitor, methyl group at C-1 of the ring does not display any inhibition
against MCF-7 cells, as explained in Table 8. The study of long term survival and sensitivity
revealed that sensitivity of HCC1806 is less than 2% and compound 90 affects the survival of
HCC1806 cells. The total pro- tein growth reduction was found after the treatment of MCF-7
and HCC1806 cells with reported target compounds. Flow cytometry study illustrates the
decrease in viable cell population in the TN (HCC1806) cell line [83].
In 2013, Soares and his team explored anti-breast cancer agents by replacement of
substituent 1H,3H-pyrrole[1,2-c]thiazole 93-96, as shown in Fig. (29). Furthermore, the SAR
study concludes that the phenyl group at C-3 and the methyl group at C-5 are responsible for
the cytotoxic activity of compound 93. It can be concluded from IC50 values that removal or
substitution of the phenyl group has a significant effect on its cytotoxic value against MCF-7
cancer cells. To evaluate anticancer activity, cancer cells were incubated for different time
intervals in DMSO solution and proliferation in cells was illustrated using the MTT assay
[84].
In 2012, Chaniyara et al. reported the synthesis of 2,3- bis(hydroxymethyl)-4H-benzo[2,1-
b]thiazoles and their bis alkyl- carbamate derivatives, as shown in Fig. (30). Furthermore, the
anti- tumor activity and DNA cross-linking ability of the compound were tested. Compound
97-99 exhibited excellent in vitro inhibition against different human solid tumor cells,
including human solid tumor breast carcinoma MX-1, lung carcinoma H1299, prostate
carcinoma PC3, COLON carcinoma HCT-116, oral carcinoma OECM1 and glioma U87
taking cisplatin as a standard reference compound, as summarized in Table 9. The antitumor
activity was tested by the xenograft model in cancer cells of mice that results in twenty
percent weight loss with approximate complete cancer cell inhibition. Cell cycle inhibition
was studied by flow cytometry and target compound 99 decreases population at a very high
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 29
ANTI CANCER DRUGS
concentra- tion of 25µM at the G1 phase after 24 hours and arrest cell cycle at G2/M phase
[85].
Thiazole Fused with Pyridine Scaffold
Thiazole [5,4-b] pyridine is a vital scaffold among the six- membered ring. In 2012, Gu and
Jin et al. prepared α-amino phos- phate derivatives 100-107 containing thiazole[5,4-b]
pyridine moi- ety, as shown in Fig. (31) and screened their anticancer activity against PC-3,
H460 cells and Bcap-37 cancer cell lines using the MTT assay. Compound 105 was found to
be the most potent inhibi- tor due to the p-fluorophenyl group, while replacement of methyl
or methoxy group results in a decrease in the activity of compounds 101 and 104. The
substituent fluoro present at the phenyl group is responsible for the significant inhibitory
activity and after removal of the fluoro group from compound 101 and 105, the inhibitory
activity decreases five times of compound 100 [86].
Thiazole Fused with the Triazine Ring
1,2,4 triazine and their bioisosterism with purines bases are found to have significant
anticancer activity. In 2019, El-Wakil et al. reported the multistep synthesis of thiazole-
hybrid triazine derivative 108, as shown in Fig. (32) and screened their anticancer activity
against a panel of 60 cancer cell lines and found that ten out of sixty cells are more sensitive
towards compound 108 with IC50 value 3.26-49.4µM. COMPARE analysis of the target
compound revealed that its antitumor inhibition was due to DNA binding simi- lar to that of
Clomesone [87].
CH2OCONHR1
CH2OCONHR1
N carbamates group responsiblefor better
cytotoxic effect than hydroxyl methyl
R2
97-99
Fig. (30). Alkylcarbamate derivative as solid tumor cell line inhibitors.
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 30
ANTI CANCER DRUGS
OH
HO
OH
S N
OH
Me S N
Ph Me
cytotoxic activity was observeddue to phenyl 94
group
93
IC = 21.5 µM
IC = 0.30 µM
HO
HO
Me
HO
MeO OMe OH
S N
N
Ph
OMe
Ph
S
95
96
IC50 = 59.9 µM
IC50 = 70.2 µM
Fig. (29). 1H, 3H pyrrole [1,2-c] thiazole derivative as anti-breast cancer agent.
Compound R1 R2 MX-1 HCT-116 H1299 PC3 OECM-1 U87
97 Et 4-F-C6H4 0.48±0.005 0.52±0.025 13.55±2.03 23.70±3.05 11.47±2.63 29.24±2.63
i
98 -Pr 4-F-C6H4 0.62±0.014 0.35±0.010 33.03±3.20 24.19±4.17 11.89±2.90 53.50±9.97
i
99 -Pr 4-Cl-C6H4 1.19±0.002 0.41±0.005 8.75±0.88 13.71±1.28 7.54±0.95 29.24±2.63
Cisplatin - - ND ND 16.53±0.90 4.7±0.66 2.44±0.53 54.53±3.33
Table 9. IC50 value against different solid tumor cells.
Fused Thiazole Linked or Tethered with other Heterocycles
Fused thiazole scaffolds fused or linked with other rings also exhibited therapeutic profile.
Many researchers have shown their interest in developing new drug candidates using fused
thiazole linked or tethered scaffold. In 2019, Prashanth et al. screened anti- cancer activity of
coumarin analogs linked with thiazole on mice leukemia cancer cells, as shown in Fig. (33).
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 31
ANTI CANCER DRUGS
Further SAR study confirmed that substitution of the methoxy group at coumarin and phenyl
fragments was responsible for the significant apoptogenic activity of compound 109. Among
the series, the most potent in- hibitory activity was evaluated using trypan blue, LDH leak
and MTT assay. The substitution of the methoxy and methyl group with hydrogen atom
decreases apoptosis. Furthermore, docking studies illustrate different interactions of target
compounds using caspase- 3, including Arg 207 or Asn 208 with hydrogen bonding with an
amide group of the target compound, a phenyl group of the target compound interacts with
the active site of caspase-3 bearing a ni- trogen atom [88].
In 2017, Keshari et al. reported one-pot-multicomponent syn- thesis of benzothiazoles
tethered with quinazoline and pyrimidine moiety. Furthermore, their anticancer study was
done on the Hep- G2 cancer cell line by sulfo rhodamine B assay. The substitution of the
indanone ring in compound 110 and tetralone ring in 111 has significantly increased the
activity against human liver cancer cells Hep-G2. In addition, the presence of the methyl and
methoxy group in the phenyl group at R1, R2, R3 and R4 group plays an important role in
inhibitory activity, as shown in Fig. (34) [89].
Al-Ghorbani et al. reported a novel series of BT tethered with the piperazine ring as a
substituent at the C-2 position, as shown in Fig. (35). Furthermore, the anticancer activity of
BT derivative 112 was performed against DLA cancer cells in vitro and the IC50 was found
25µM, 22.6µM, and 23µM in trypan blue, MTT and LDH assays, respectively. SAR study of
the BT derivative reveals that the Bromine group at the phenyl ring is responsible for
angiogene- sis inhibition. The tumor inhibitory potential of the target was ob- served due to
the capture of the angiogenesis process on rVEGF165 using Chorio Allanotoic Membrane
(CAM) assay [90].
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 32
ANTI CANCER DRUGS
CONCLUSION
Heterocyclic compounds possess significance in medicinal chemistry as anticancer agents as
heterocyclic compounds occupy an approximate total of two-third anticancer drugs approved
by the FDA. Fused thiazole is an important core nucleus present in many biologically active
compounds, clinical trials drugs and FDA ap- proved drugs. These now become interesting
candidates for chem- ists and researchers due to their wide broad-spectrum properties. we
have comprised various synthetic approaches for fused thiazoles and their anticancer
properties in this review. Furthermore, various thiazole fused compounds including
benzothiazole, imidazo thia zole, pyrrolopyrimidines, thiazoles fused with pyridine, thiazole
tethered with steroids have explored with their anticancer activity. Numerous
multicomponent reactions are reported, which are rele- vant for the synthesis of a plethora of
thiazole tethered/fused scaf- folds as a template for biologically active pharmacophore.
Several types of cancer inhibitors, including human breast, colon, liver, prostate, lung
pancreatic ovarian, renal, are explored, which is a fruitful approach for future drug
development, aimed to combat cancer. SAR and docking study reveals that though the
anticancer activity of fused thiazole scaffold containing compounds depends on various
substituents present on the ring and different types of binding interactions with the target
compound, it is not always logi-cal and thus difficult to interpret. This review included
several tar- get molecules with their SAR insight, which might provide a lead to further
development of more potent biological active drugs contain- ing fused thiazoles moieties.
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 33
ANTI CANCER DRUGS
References
1. 1.Benvenga S. 2005 Peripheral hormone metabolism thyroid hormone transport
proteins and the physiology of hormone binding, In: Werner&Ingbar’s The Thyroid a
Fundamental and clinical Text, Braverman, LE.&Utiger, RD., 97 105 ), Lippincott
Williams&Wilkins Company, 0-7817-5047-4, Philadelphia.
2. 2.Bursuk E. Gulcur H. Ercan M. 2010 The significance of body impedance and blood
viscosity measurements in thyroid diseases, Proceedings of Biomedical Engineering
Meeting (BIYOMUT), 15th National, 978-1-4244-6380-0, Antalya, April 2010
(http://ieeexplore.ieee.org/xpls/abs_all.jsp?arnumber=5479828&tag=1).
3. 3.Di Lauro R. De Felice M. 2001 Bas
4. ic Physiology anatomy development, In: Endocrinology, DeGroot, LJ.&Jameson,
JL., 1268 1275 ), W.B. Saunders Company, 0-7216-7840-8, Philadelphia.
5. 4.Dillmann W. H. 2004 The thyroid, In: Cecil Textbook of Medicine, Goldman,
L.&Ausrello, D., 1391 1411 ), Saunders, Philadelphia.
6. 5.Dunn J. T. 2001 Biosynthesis and secretion of thyroid hormones, In: Endocrinology,
DeGroort, LJ.,&Jameson, JL., 1290 1298 ), W.B. Saunders Company, 0-7216-7840-8,
Philadelphia.
7. 6.Ganong W. F. 1997 Review of Medical Physiology (eighteenth edition),
Appleton&Lange, 0- 8385-8443-8, Stamford.
8. 7.Guyton A. C. Hall J. E. 2006 Textbook of Medical Physiology (eleventh edition),
Elsevier Sanders, 0-7216-0240-1, Philadelphia.
9. 8.Jameson J. L. Weetman A. P. 2010 Disorders of the thyroid gland, In: Harrison’s
Endocrinology, Jameson, JL., 62 69 ), The McGraw-Hill Companies, Inc., 978-0-07-
174147-7, New York.
10. 9.Larsen P. R. Davies T. F. Schlumberger M. J. Hay I. D. 2003 Thyroid physiology
and diagnostic evaluation of patients with thyroid disorders, In: Williams Textbook of
Endocrinology, Larsen, PR., Kronenberg, HM., Melmed, S.&Polonsky,
KS., 331 353 ), Saunders, 0-7216-9184-6, Philadelphia.
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 34
ANTI CANCER DRUGS
11. 10.Lo Presti. J. S. Singer P. A. 1997 Physiology of thyroid hormone synthesis,
secretion, and transport, In: Thyroid Disease Endocrinology, Surger, Nuclear
Medicine and Radiotherapy. Falk, SA, 29 39 ), Lippincott-Raven Publishers, 0-397-
51705-X, Philadelphia.
12. 11.Mc Gregor A. M. 1996 The thyroid gland and disorders of thyroid function,
In: Oxford Fextbook of Medicine, Weatherall, DJ., Ledingham, JGG. & Warrell,
DA, 1603 1621 ), Oxford University Press, 0-19-262707-4, Oxford, 2
13. 12.Reed L. Pangaro L. N. 1995 Physiology of the thyroid gland I: synthesis and
release, iodine metabolism, and binding and transport, In: Principles and Practice of
Endocrinology and Metabolism, Becher, KL., 285 291 ), J.B. Lippincott Company, 0-
397-51404-2, Philadelphia.
14. 13.Santiseban P. 2005 Development and anatomy of the hypothalamic- pituitary-
thyroid axis, In: Werner&Ingbar’s The Thyroid a Fundamental and Clinical Text,
Braverman, LE.,&Utiger, RD., 8 23 ), Lippincot Williams&Wilkins Company, 0-
7817-5047-4, Philadelphia.
15. 14.Scanlon M. F. 2001 Thyrothropin releasing hormone and thyrothropin stimulating
hormone, In: Endocrinology, DeGroot, LJ.&Jameson, JL., 1279 1286 ), W.B.
Saunders Company, 0-7216-7840-8, Philadelphia.
16. 15.Snell R. S. 1995 Clinical Anatomy for my students (fifth edition), Little, Brown
and Company, 0-316-80135-6, Boston.
17. 16.Usala S. J. 1995 Physiology of the thyroid gland II: reseptors, postreceptor events,
and hormone resistance syndromes, In: Principle and Practice of Endocrinology and
Metabolism, Becker, KL., 292 298 ), J.B. Lippincott Company, 0-397-51404-2,
Philadelphia.
18. 17.Utiger R. D. 1997 Disorders of the thyroid gland, In: Textbook of Intecnal
Medrane, Kelley, WN., 2204 2219 ), Lippincott- Raven Publishers, 0-397-51540-5,
Philadelphia. . Andrews, Gavin P., Thomas P. Laverty, and David S. Jones.
"Mucoadhesive Polymeric Platforms for Controlled Drug Delivery." European
Journal of Pharmaceutics and Biopharmaceutics 71.3 (2009): 505-18. Print.
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 35
ANTI CANCER DRUGS
19. 2. Asane, GS, et al., "Polymers for Mucoadhesive Drug Delivery System: A Current
Status." Drug development and industrial pharmacy 34.11 (2008): 1246-66. Print.
20. 3. Chanburee, Sanipon, and Waree Tiyaboonchai. "Mucoadhesive Nanostructured
Lipid Carriers (NLCs) as Potential Carriers for Improving Oral Delivery of
Curcumin." Drug development and industrial pharmacy 43.3 (2017): 432-40. Print.
21. 4. Dodou, Dimitra, Paul Breedveld, and Peter A. Wieringa. "Mucoadhesives in the
Gastrointestinal Tract:
22. 5. Jain, Shashank, et al., "Formulation and Rheological Evaluation of Ethosome-
Loaded Carbopol Hydrogel for Transdermal Application." Drug development and
industrial pharmacy 42.8 (2016): 1315-24. Print.
23. 6. Jain, Shashank, et al., "Quality by Design Approach for Formulation, Evaluation
and Statistical Optimization of Diclofenac-Loaded Ethosomes via Transdermal
Route." Pharmaceutical development and technology 20.4 (2015): 473-89. Print.
24. 7. Jain, Shashank, et al., "Recent Advances in Lipid-Based Vesicles and Particulate
Carriers for Topical and Transdermal Application." Journal of pharmaceutical
sciences (2016) Print.
25. 8. Luo, Yangchao, et al., "Solid Lipid Nanoparticles for Oral Drug Delivery: Chitosan
Coating Improves Stability, Controlled Delivery, Mucoadhesion and Cellular
Uptake." Carbohydrate Polymers 122 (2015): 221-9. Print.
26. 9. Mazzarino, Letícia, et al., "Xyloglucan‐block‐Poly (ϵ‐Caprolactone) Copolymer
Nanoparticles Coated with Chitosan as Biocompatible Mucoadhesive Drug Delivery
System." Macromolecular bioscience 14.5 (2014): 709-19. Print.
27. 10. Brown, A.J. On an Acetic Ferment which form Cellulose. J. Chem. Soc. 1986, 49,
172–187. [CrossRef]
28. 11. Mohite, B.V.; Patil, S.V. A novel biomaterial: Bacterial cellulose and its new era
applications. Biotechnol. Appl. Biochem. 2014, 61, 101–110. [CrossRef]
29. 12. Czaja, W.K.; Young, D.J.; Kawecki, M.; Brown, R.M. The Future Prospects of
Microbial Cellulose in Biomedical Applications. Biomacromolecules 2007, 8, 1–12.
[CrossRef]
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 36
ANTI CANCER DRUGS
30. 13. Hestrin, S.; Schramm, M. Synthesis of cellulose by Acetobacter xylinum. 2.
Preparation of freeze-dried cells capable of polymerizing glucose to cellulose.
Biochem. J. 1954, 58, 345–352. [CrossRef]
31. 14. Lestari, P.; Elfrida, N.; Suryani, A.; Suryadi, Y. Study on the Production of
Bacterial Cellulose from Acetobacter Xylinum Using Agro—Waste. Jordan J. Biol.
Sci. 2014, 7, 75–80. [CrossRef]
32. 15. Saha, N.; Vyroubal, R.; Sáha, P. Apple Juice: An alternative feed-stock to
enhance the production of Bacterial Nano Cellulose. In Proceedings of the 2nd
International Symposium on Bacterial Nanocellulose, Gda ´nsk, Poland, 9–11
September 2015.
33. 16. Zandraa, O.; Saha, N.; Shimoga, G.D.; Palem, R.R.; Saha, P. Bacterial Cellulose,
an excellent biobased polymer produced from Apple, Book of Abstract Juice. In
Proceedings of the 9th International Conference on Modification, Degradation and
Stabilization of Polymers, Krakow, Poland, 4–8 September 2016.
34. 17. Bandopadhyay, S.; Saha, N.; Zandraa, O.; Saha, P. Bacterial cellulose from apple
juice—A polysaccharide based bioadditive for sustainable food packaging, Abstract
Book, 35–36. In Proceedings of the 5th EPNOE International Polysaccharide
Conference, Jena, Germany, 20–24 August 2017.
35. 18. MohammadKazemi, F.; Azin, M.; Ashori, A. Production of bacterial cellulose
using different carbon sources and culture media. Carbohydr. Polym. 2015, 117, 518–
523. [CrossRef]
36. 19. Gardner, J.D.; Oporto, S.G.; Mills, R.; Samir, A.S.A.M. Adhesion and surface
Issues in Cellulose and Nanaocellulose. J. Adhes. Sci. Technol. 2008, 22, 545–567.
[CrossRef]
37. 20. Oner, E.T.; Hernández, L.; Combie, J. Review of Levan polysaccharide: From a
century of past experiences to future prospects. Biotechnol. Adv. 2016, 34, 827–844.
[CrossRef]
BHOPAL INSTITUTE OF TECHNOLOGY AND SCIENCE-PHARMACY Page 37
You might also like
- Aurora Kinase Inhibitors As Anticancer AgentsDocument17 pagesAurora Kinase Inhibitors As Anticancer Agentsrohitgautam25No ratings yet
- Chapter-3 Synthesis, Characterization of Some Novel Chromeno Oxadiazole DerivativesDocument43 pagesChapter-3 Synthesis, Characterization of Some Novel Chromeno Oxadiazole DerivativesMadalina GhitaNo ratings yet
- Chemistry SynopsisDocument7 pagesChemistry SynopsisDr Prashant Shihora100% (1)
- Heterocyclic Chemistry Problem Set SynthesesDocument1 pageHeterocyclic Chemistry Problem Set SynthesesaNo ratings yet
- Synthetic Approaches To The 2010-2014 New AgrochemicalsDocument65 pagesSynthetic Approaches To The 2010-2014 New AgrochemicalsprashantNo ratings yet
- Antibiotik Betha LaktamDocument18 pagesAntibiotik Betha LaktamdmujahidinNo ratings yet
- Thiosemicarbazides Synthesis andDocument32 pagesThiosemicarbazides Synthesis andAnis BouchamaNo ratings yet
- Biochemistry (Digestion) (269-272)Document4 pagesBiochemistry (Digestion) (269-272)Handika SaputraNo ratings yet
- ConazolesDocument60 pagesConazolesJulii TrochezNo ratings yet
- Novel Heterocyclyl Linked Benzaldehydes and Anilines for Biologically Active CompoundsDocument7 pagesNovel Heterocyclyl Linked Benzaldehydes and Anilines for Biologically Active CompoundsDr. Bharat SutharNo ratings yet
- Substrate-B Synthesis Route OptimizationDocument2 pagesSubstrate-B Synthesis Route OptimizationsauronsauronNo ratings yet
- Patrick Ch22 p3Document12 pagesPatrick Ch22 p3Oxy GenNo ratings yet
- FDA Sulphur DrugsDocument34 pagesFDA Sulphur Drugspraveen kumarNo ratings yet
- Synthesis, Characterization and Bioassay of Novel Substituted 1 - (3 - (1,3-Thiazol-2-Yl) Phenyl) - 5-OxopyrrolidinesDocument20 pagesSynthesis, Characterization and Bioassay of Novel Substituted 1 - (3 - (1,3-Thiazol-2-Yl) Phenyl) - 5-OxopyrrolidinesAna-Maria CiobotaruNo ratings yet
- Only MethodologyDocument6 pagesOnly MethodologySidu BhosleNo ratings yet
- Pro. Dr. Muhammad Ashraf Chairman Department of Pharmacology and Toxicology, UVAS, LahoreDocument29 pagesPro. Dr. Muhammad Ashraf Chairman Department of Pharmacology and Toxicology, UVAS, LahoreMuhammad Shahid BilalNo ratings yet
- Sintesis de IndolesDocument14 pagesSintesis de IndolesLili AmadorNo ratings yet
- 3COPY Der Pharma Chemica Lamiaa EtalDocument11 pages3COPY Der Pharma Chemica Lamiaa EtalLaila Abou zeidNo ratings yet
- KJM5320 Kap 2Document24 pagesKJM5320 Kap 2Raquel da Silva JustinoNo ratings yet
- Ceftriaxone Sodium: + NaohDocument2 pagesCeftriaxone Sodium: + NaohBilal MasoodNo ratings yet
- Analysis of US FDA Approved Drugs Containing Sulfur Atoms: Kevin A. Scott Jon T. NjardarsonDocument34 pagesAnalysis of US FDA Approved Drugs Containing Sulfur Atoms: Kevin A. Scott Jon T. NjardarsonRead WhiteNo ratings yet
- Practical FCHG412 Prodrugs: Module Outcomes For The PracticalDocument14 pagesPractical FCHG412 Prodrugs: Module Outcomes For The PracticalGreg RalphNo ratings yet
- Molecules: Recent Developments and Biological Activities of N-Substituted Carbazole Derivatives: A ReviewDocument22 pagesMolecules: Recent Developments and Biological Activities of N-Substituted Carbazole Derivatives: A Reviewsatheeshpharma6No ratings yet
- Matheus Síntese de Produto Natural 439Document4 pagesMatheus Síntese de Produto Natural 439MATHEUS PHILYPI ALVES VAZNo ratings yet
- Heterocycles chemistry guideDocument23 pagesHeterocycles chemistry guideRamsha AwanNo ratings yet
- General Molecular Principle in Drug Design: Dr. Rudi Hendra Sy. M.SC., AptDocument27 pagesGeneral Molecular Principle in Drug Design: Dr. Rudi Hendra Sy. M.SC., AptRifqi Ahmad IrzamiNo ratings yet
- Biology_30__Molecular_Genetics_Notes_Review_2021.docDocument15 pagesBiology_30__Molecular_Genetics_Notes_Review_2021.docsherlynmamac98No ratings yet
- PT LIPO - GLDocument2 pagesPT LIPO - GLGiulia LodigianiNo ratings yet
- Figure 1-NITROSAMINES - CONTAMINATIONDocument1 pageFigure 1-NITROSAMINES - CONTAMINATIONد. وليد الشريفNo ratings yet
- Zeocin ManDocument24 pagesZeocin ManThaís Paiva Porto de SouzaNo ratings yet
- Drug Design: Functional Groups / Pharmacological ActivityDocument24 pagesDrug Design: Functional Groups / Pharmacological ActivityIoana Mirela VasincuNo ratings yet
- Lecture Notes Chem 51A S. King: O H OH H HO HO H H H CH OHDocument41 pagesLecture Notes Chem 51A S. King: O H OH H HO HO H H H CH OHTamiaNo ratings yet
- Fisher 2012Document8 pagesFisher 2012good devilNo ratings yet
- HNR, T3P/E Toac, Huning'S Base, CH CL Lioh, Thf/H2ODocument1 pageHNR, T3P/E Toac, Huning'S Base, CH CL Lioh, Thf/H2OJuan Antonio de la RosaNo ratings yet
- Lectures in Heterocyclic ChemistryDocument130 pagesLectures in Heterocyclic ChemistryNorman FerdinalNo ratings yet
- Unit 3 QuestionsDocument18 pagesUnit 3 QuestionsLynd TaylorNo ratings yet
- Novel Benzimidazole-Linked Pyrazolines as Potential Antimicrobial AgentsDocument9 pagesNovel Benzimidazole-Linked Pyrazolines as Potential Antimicrobial Agentsg20kpNo ratings yet
- 3,5-Bis (2-Indolyl) Pyridine and 3 - ( (2-Indolyl) - 5-Phenyl) Pyrid PDFDocument22 pages3,5-Bis (2-Indolyl) Pyridine and 3 - ( (2-Indolyl) - 5-Phenyl) Pyrid PDFpaiaravindNo ratings yet
- 2016 Delt Paper Revision Accepted SupplementaryDocument11 pages2016 Delt Paper Revision Accepted SupplementarypufuNo ratings yet
- Bio IngDocument29 pagesBio Ingsudipta88No ratings yet
- Nucleac Acid Mid Bio IIDocument47 pagesNucleac Acid Mid Bio IIseada JemalNo ratings yet
- Drug Design: Functional Groups / Pharmacological ActivityDocument24 pagesDrug Design: Functional Groups / Pharmacological ActivityMahniar SinagaNo ratings yet
- Medicinal Chemistry & Drug Discovery: Dr. Peter Wipf Department of Chemistry University of PittsburghDocument69 pagesMedicinal Chemistry & Drug Discovery: Dr. Peter Wipf Department of Chemistry University of PittsburghKhushi KhanNo ratings yet
- N-Demethylation of Alkaloids: A Key Transformation in Drug SynthesisDocument13 pagesN-Demethylation of Alkaloids: A Key Transformation in Drug SynthesisPhuongNo ratings yet
- Answers ch10Document6 pagesAnswers ch10김아진No ratings yet
- molecules-20-03821Document20 pagesmolecules-20-03821putryapurnomo.21No ratings yet
- E12 AtqDocument5 pagesE12 AtqCharlene InaoNo ratings yet
- Fenol 4-Metil Fenol: HO OH CHDocument1 pageFenol 4-Metil Fenol: HO OH CHrohman cooyNo ratings yet
- Fenol 4-Metil Fenol: HO OH CHDocument1 pageFenol 4-Metil Fenol: HO OH CHrohman cooyNo ratings yet
- Keys To The Problems (2021/02/13) : Blue LEDDocument4 pagesKeys To The Problems (2021/02/13) : Blue LEDHuân TrầnNo ratings yet
- Thia Zoli Dined I OnesDocument6 pagesThia Zoli Dined I OnesSoumyadeep MondalNo ratings yet
- Antineoplastics 22Document50 pagesAntineoplastics 22ahmed montaserNo ratings yet
- Chapter Solutions 14Document6 pagesChapter Solutions 14luizdrNo ratings yet
- Chemistry of FormazanDocument36 pagesChemistry of FormazanEsteban ArayaNo ratings yet
- Macrolide Antibiotics: Binding Site, Mechanism of Action, ResistanceDocument15 pagesMacrolide Antibiotics: Binding Site, Mechanism of Action, ResistancevitulNo ratings yet
- 2006-CHM6108 - L5L6 HandoutDocument9 pages2006-CHM6108 - L5L6 Handoutaidar.seralinNo ratings yet
- Antisense Oligonucleotide Biotechnology, Applications and FutureDocument29 pagesAntisense Oligonucleotide Biotechnology, Applications and FuturesurojitarpitaNo ratings yet
- DNA and Animal Classification: Year Ten StudentsDocument20 pagesDNA and Animal Classification: Year Ten StudentsCamille Sison-AlmirolNo ratings yet
- Andrew Fant University of North Carolina Eshelman School of PharmacyDocument24 pagesAndrew Fant University of North Carolina Eshelman School of PharmacyuncesopNo ratings yet
- Stabilitas Ampi-Lipid EmulsDocument12 pagesStabilitas Ampi-Lipid EmulsrosianaNo ratings yet
- GPAT Pharmaceutics SyllabusDocument5 pagesGPAT Pharmaceutics Syllabuskumar HarshNo ratings yet
- Chapter - 2 Hospital PharmacyDocument33 pagesChapter - 2 Hospital PharmacyPallavi PatilNo ratings yet
- Pharmacology Calculations: Solving Dosage Problems Step-by-StepDocument54 pagesPharmacology Calculations: Solving Dosage Problems Step-by-StepAryan KhanNo ratings yet
- Mapa de Controlo de Abril Farmacia 1 de MaioDocument7 pagesMapa de Controlo de Abril Farmacia 1 de Maiolusineide rosarioNo ratings yet
- Contoh LASADocument1 pageContoh LASAninananaNo ratings yet
- Forensic pharmacy & the role of forensic pharmacistsDocument16 pagesForensic pharmacy & the role of forensic pharmacistsJaved IqbalNo ratings yet
- Product Specification: Product Description: Product CodeDocument1 pageProduct Specification: Product Description: Product CodeUmar AliNo ratings yet
- Batuk - Pilek Harga Jumlah Barang: Satuan Masuk Terjual SisaDocument7 pagesBatuk - Pilek Harga Jumlah Barang: Satuan Masuk Terjual Sisadoni januarindraNo ratings yet
- Probenecid Drug StudyDocument1 pageProbenecid Drug StudykyawNo ratings yet
- PMC Buys Isochem - Generics No 334 20171205Document16 pagesPMC Buys Isochem - Generics No 334 20171205Echo WackoNo ratings yet
- Lecture-2 HISTORY OF PHARMACY Regarding The Muslim ScientistDocument10 pagesLecture-2 HISTORY OF PHARMACY Regarding The Muslim ScientistHuda Qureshi Hashmi QureshiNo ratings yet
- PowerPoint PresentationSNLDocument6 pagesPowerPoint PresentationSNLAfifah N AhmedNo ratings yet
- Holdings Daily Us en GMFDocument84 pagesHoldings Daily Us en GMFstarNo ratings yet
- Over The Counter Medicine For Erectile DysfunctionlftqgDocument3 pagesOver The Counter Medicine For Erectile Dysfunctionlftqgpeapike0No ratings yet
- High Performance Thin Layer Chromatographic Method With Densitometry Analysis For Determination of Rivaroxaban From Its Tablet Dosage FormDocument4 pagesHigh Performance Thin Layer Chromatographic Method With Densitometry Analysis For Determination of Rivaroxaban From Its Tablet Dosage FormPinak PatelNo ratings yet
- Principle of Drug ActionDocument4 pagesPrinciple of Drug ActionChristopher jan LegaspiNo ratings yet
- Magic Mushroom Spores - Where To Buy Psilocybin Spo - 240105 - 183240Document24 pagesMagic Mushroom Spores - Where To Buy Psilocybin Spo - 240105 - 183240coreeastcapitalNo ratings yet
- Clinical Toxicology: Unit I: General Principles of ToxicologyDocument11 pagesClinical Toxicology: Unit I: General Principles of ToxicologyAnnaNo ratings yet
- Drug Alert List of Oct - 2022Document11 pagesDrug Alert List of Oct - 2022Gopal RaoNo ratings yet
- Azole Antifungals HandoutDocument8 pagesAzole Antifungals Handoutapi-648714317No ratings yet
- Types, Manufacture, Formulation of Capsules 1Document26 pagesTypes, Manufacture, Formulation of Capsules 1chill streamNo ratings yet
- Encube Corporate PresentationDocument19 pagesEncube Corporate PresentationAnnu KambleNo ratings yet
- SCIENCE6 - Q1 - MOD9 - Benefits of Separating MixturesDocument21 pagesSCIENCE6 - Q1 - MOD9 - Benefits of Separating MixturesSophia Magdayao100% (4)
- Elixirs - Hydroalcoholic SolutionsDocument6 pagesElixirs - Hydroalcoholic SolutionsVoice Over Verses100% (1)
- Bronchodilation Effects of Kalachuchi Leaves (Plumeria Induced BronchoconstrictionDocument15 pagesBronchodilation Effects of Kalachuchi Leaves (Plumeria Induced BronchoconstrictionLenny Mae JagosNo ratings yet
- Module 5 ActivityDocument12 pagesModule 5 ActivityEricka Mae AbreaNo ratings yet
- Role of pharmacists in small hospitals and nursing homesDocument22 pagesRole of pharmacists in small hospitals and nursing homesKinza KhanNo ratings yet
- Acyclovir: An SEO-Optimized TitleDocument3 pagesAcyclovir: An SEO-Optimized TitleRahma SantosoNo ratings yet
- Strategic Management of Biocon India GroupDocument6 pagesStrategic Management of Biocon India GroupKisan Sahu100% (1)