You might also like
- Vinyl Chloride Monomer/Ethylene Dichloride (VCM/EDC)Document10 pagesVinyl Chloride Monomer/Ethylene Dichloride (VCM/EDC)PuLung Sambadha100% (1)
- En NB SkillsAssessment-Student Exam (Martin)Document10 pagesEn NB SkillsAssessment-Student Exam (Martin)Marthin Charles Hutagalung50% (4)
- Employee Motivation Literature ReviewDocument18 pagesEmployee Motivation Literature Reviewapi-297379255No ratings yet
- Statistical Molecular Thermodynamics: Christopher J. CramerDocument7 pagesStatistical Molecular Thermodynamics: Christopher J. CramerLuciano Sánchez AramburuNo ratings yet
- CH 11Document14 pagesCH 11hirenpatel_universalNo ratings yet
- CHEM443 Fall 2014 Oldquiz1 SolutionDocument4 pagesCHEM443 Fall 2014 Oldquiz1 SolutionLin Xian XingNo ratings yet
- 2-Fundamental Property RelationsDocument24 pages2-Fundamental Property RelationsIkNo ratings yet
- Chapter 2 BDocument11 pagesChapter 2 BStefanPerendijaNo ratings yet
- ch05 PDFDocument83 pagesch05 PDFIsabelHutterNo ratings yet
- Calculation of Properties of Pure FluidsDocument143 pagesCalculation of Properties of Pure FluidsArdila Hayu TiwikramaNo ratings yet
- CET I 4. Properties of Fluid 2020 SentDocument22 pagesCET I 4. Properties of Fluid 2020 Sent5fdt78kgscNo ratings yet
- Assignment - 2Document20 pagesAssignment - 2comedy worldNo ratings yet
- Jeans Instability and Gravitational Collapse: Fluid Dynamics 101Document7 pagesJeans Instability and Gravitational Collapse: Fluid Dynamics 101SDasNo ratings yet
- Section 2D. State Functions and Exact DifferentialsDocument4 pagesSection 2D. State Functions and Exact DifferentialsAkib ImtihanNo ratings yet
- Introduction To The Equilibrium PhaseDocument8 pagesIntroduction To The Equilibrium PhaseJksgNo ratings yet
- Compressibility PDFDocument2 pagesCompressibility PDFJuan Daniel CabreraNo ratings yet
- Chapter 3 Bernoulli Equation: 3.1 Flow Patterns: Streamlines, Pathlines, StreaklinesDocument25 pagesChapter 3 Bernoulli Equation: 3.1 Flow Patterns: Streamlines, Pathlines, Streaklinesengineer63No ratings yet
- Exer E 2Document3 pagesExer E 2prathameshkawtikwar31No ratings yet
- Finite Volume Method For One DimensionalDocument26 pagesFinite Volume Method For One Dimensionalscience1990No ratings yet
- Lecture 3Document19 pagesLecture 3Suvrasoumya MohantyNo ratings yet
- ZCT 214 - Lecture - Chapter6Document43 pagesZCT 214 - Lecture - Chapter6Nur Athirah KhairinaNo ratings yet
- Module 3Document38 pagesModule 3Rutu PatilNo ratings yet
- Assignment - 1, 2, 3 (Save)Document60 pagesAssignment - 1, 2, 3 (Save)comedy worldNo ratings yet
- Chapter 6: Thermodynamic Properties of Real Fluids The Four Fundamental Property RelationsDocument51 pagesChapter 6: Thermodynamic Properties of Real Fluids The Four Fundamental Property RelationsBilal AhmedNo ratings yet
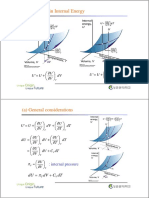
- 2.11 Changes in Internal Energy: DV V U U U DT T U U UDocument6 pages2.11 Changes in Internal Energy: DV V U U U DT T U U UHyeon Chang NoNo ratings yet
- Homework Problem Set 4 Solutions: Chemistry 360 Dr. Jean M. StandardDocument10 pagesHomework Problem Set 4 Solutions: Chemistry 360 Dr. Jean M. Standardisaac wekesaNo ratings yet
- Summary Chapter2Document4 pagesSummary Chapter2blalllNo ratings yet
- Homework 1Document7 pagesHomework 1jhony95No ratings yet
- Report-Open-Hydraulics-Energy EquationDocument8 pagesReport-Open-Hydraulics-Energy EquationJohn Eric CabañaNo ratings yet
- Maxwell EquationDocument2 pagesMaxwell Equation黃士軒No ratings yet
- The Laplacian in Curvilinear Coordinates - The Full Story PDFDocument77 pagesThe Laplacian in Curvilinear Coordinates - The Full Story PDFKARAN NADHNo ratings yet
- l6 ThermoDocument68 pagesl6 ThermoJahnavi RameshNo ratings yet
- Gurney-Lurie ChartsDocument90 pagesGurney-Lurie ChartsMuhammad Nur JumadilNo ratings yet
- Introductory Chemical Engineering Thermodynamics: by J.R. Elliott and C.T. LiraDocument9 pagesIntroductory Chemical Engineering Thermodynamics: by J.R. Elliott and C.T. LiraAman SinghNo ratings yet
- Felix Termodinamica Quimica ch05Document83 pagesFelix Termodinamica Quimica ch05Amilcar Pereira CardosoNo ratings yet
- BasicDeriv PDFDocument1 pageBasicDeriv PDFFrederick HeisenbergNo ratings yet
- BasicDeriv PDFDocument1 pageBasicDeriv PDFsenahidNo ratings yet
- BasicDeriv PDFDocument1 pageBasicDeriv PDFFrederick HeisenbergNo ratings yet
- BasicDeriv PDFDocument1 pageBasicDeriv PDFRicky Putra SiregarNo ratings yet
- BasicDeriv PDFDocument1 pageBasicDeriv PDFFrederick HeisenbergNo ratings yet
- BasicDeriv PDFDocument1 pageBasicDeriv PDFRicky Putra SiregarNo ratings yet
- Du Tds PDV DH Tds VDP Da SDT PDV DG SDT VDP: Chapter 11 SummaryDocument4 pagesDu Tds PDV DH Tds VDP Da SDT PDV DG SDT VDP: Chapter 11 SummaryGitanjali TomarNo ratings yet
- 3 The First Law: The Machinery: Solutions To ExercisesDocument13 pages3 The First Law: The Machinery: Solutions To ExercisesRenan Gustavo PazNo ratings yet
- Physics - TDS EquationsDocument4 pagesPhysics - TDS EquationsAmolNo ratings yet
- Homework Week 8Document12 pagesHomework Week 8216435964No ratings yet
- 5-Summary For Lectures 1,2,3,4Document17 pages5-Summary For Lectures 1,2,3,4losblancos1No ratings yet
- Continuity Equation / Conservation of MassDocument32 pagesContinuity Equation / Conservation of MassmanigandanNo ratings yet
- Module-5: Constitutive Relations: Lecture-40: Thermomechanics of Incompressible FluidsDocument5 pagesModule-5: Constitutive Relations: Lecture-40: Thermomechanics of Incompressible FluidsKevin Henoc AYINo ratings yet
- HydronoteDocument7 pagesHydronoteBaishali SurNo ratings yet
- Heat Equation Ajit KumarDocument3 pagesHeat Equation Ajit KumarAjit KumarNo ratings yet
- Hand Outs CHE201 Physical ChemistryDocument73 pagesHand Outs CHE201 Physical ChemistrySHER AfghanNo ratings yet
- 5.60 Thermodynamics & Kinetics: Mit OpencoursewareDocument5 pages5.60 Thermodynamics & Kinetics: Mit OpencoursewarecaptainhassNo ratings yet
- Lecture 2Document22 pagesLecture 2بوبي بابيNo ratings yet
- 3A5 Examples Paper 2Document10 pages3A5 Examples Paper 2Pablo PeñasNo ratings yet
- EMG - 2307 Chapter 4 - 2021Document9 pagesEMG - 2307 Chapter 4 - 2021VictoriaNo ratings yet
- 5.60 Thermodynamics & Kinetics: Mit OpencoursewareDocument8 pages5.60 Thermodynamics & Kinetics: Mit OpencoursewarecaptainhassNo ratings yet
- FM Mod3@Azdocuments - inDocument29 pagesFM Mod3@Azdocuments - inVivek Thakur SujanianNo ratings yet
- TermodinâmicaDocument3 pagesTermodinâmicathiagopoetarnNo ratings yet
- Lecture 10 - Transmission Lines: X X y yDocument8 pagesLecture 10 - Transmission Lines: X X y ysamer saeedNo ratings yet
- Combining The 1 and 2 Laws: Du DQ + DW T Ds - P DV Ds DQ /T Du/T - DW/T Ds Du/T + P/T DV Du Tds - PDVDocument12 pagesCombining The 1 and 2 Laws: Du DQ + DW T Ds - P DV Ds DQ /T Du/T - DW/T Ds Du/T + P/T DV Du Tds - PDVDtf6969No ratings yet
- HW5 SolDocument3 pagesHW5 SoloppipxNo ratings yet
- Lecture16 Equations IsothermalSystems 3Document11 pagesLecture16 Equations IsothermalSystems 3shubhamNo ratings yet
- The Spectral Theory of Toeplitz Operators. (AM-99), Volume 99From EverandThe Spectral Theory of Toeplitz Operators. (AM-99), Volume 99No ratings yet
- Statistical Molecular Thermodynamics: Christopher J. CramerDocument4 pagesStatistical Molecular Thermodynamics: Christopher J. CramerLuciano Sánchez AramburuNo ratings yet
- Statistical Molecular Thermodynamics: Christopher J. CramerDocument7 pagesStatistical Molecular Thermodynamics: Christopher J. CramerLuciano Sánchez AramburuNo ratings yet
- Statistical Molecular Thermodynamics: Christopher J. CramerDocument7 pagesStatistical Molecular Thermodynamics: Christopher J. CramerLuciano Sánchez AramburuNo ratings yet
- Statistical Molecular Thermodynamics: Christopher J. CramerDocument8 pagesStatistical Molecular Thermodynamics: Christopher J. CramerLuciano Sánchez AramburuNo ratings yet
- Statistical Molecular Thermodynamics: Christopher J. CramerDocument7 pagesStatistical Molecular Thermodynamics: Christopher J. CramerLuciano Sánchez AramburuNo ratings yet
- 2000 The Application of The Gurson Tvergaard Model inDocument9 pages2000 The Application of The Gurson Tvergaard Model inLuciano Sánchez AramburuNo ratings yet
- Statistical Molecular Thermodynamics: Christopher J. CramerDocument7 pagesStatistical Molecular Thermodynamics: Christopher J. CramerLuciano Sánchez AramburuNo ratings yet
- Statistical Molecular Thermodynamics: Christopher J. CramerDocument5 pagesStatistical Molecular Thermodynamics: Christopher J. CramerLuciano Sánchez AramburuNo ratings yet
- 0460 w13 QP 22 PDFDocument16 pages0460 w13 QP 22 PDFLuciano Sánchez AramburuNo ratings yet
- 0460 w13 QP 42 PDFDocument16 pages0460 w13 QP 42 PDFLuciano Sánchez AramburuNo ratings yet
- 0460 w13 Ms 42 PDFDocument5 pages0460 w13 Ms 42 PDFLuciano Sánchez AramburuNo ratings yet
- 0460 w13 Ms 22 PDFDocument6 pages0460 w13 Ms 22 PDFLuciano Sánchez AramburuNo ratings yet
- 0460 s13 Ms 12Document14 pages0460 s13 Ms 12Luciano Sánchez AramburuNo ratings yet
- Graduation Paper TUDelft DPdeBruijn 4036549 RepositoryDocument197 pagesGraduation Paper TUDelft DPdeBruijn 4036549 RepositoryErmal SpahiuNo ratings yet
- AGRD04B-15 Guide To Road Design Part 4B Roundabouts Ed3.1Document97 pagesAGRD04B-15 Guide To Road Design Part 4B Roundabouts Ed3.1fbturaNo ratings yet
- Managing Behavioral Changes in An OrganizationDocument15 pagesManaging Behavioral Changes in An OrganizationLemuel AyingNo ratings yet
- RT130 RCI Calibration ProcessDocument6 pagesRT130 RCI Calibration ProcessAlan Martin De La Cruz VazquezNo ratings yet
- Faculty Manuals Pages 80 - 90Document10 pagesFaculty Manuals Pages 80 - 90Maria Faye MarianoNo ratings yet
- AluminiumDocument31 pagesAluminiumsamuelNo ratings yet
- Chapter 5 QuizDocument6 pagesChapter 5 QuizArunNo ratings yet
- TriplexerDocument1 pageTriplexerKarim FaisalNo ratings yet
- Canons 18 19Document36 pagesCanons 18 19Shawn Lee100% (1)
- Making Sense of VoluDocument44 pagesMaking Sense of VoluImprovingSupportNo ratings yet
- Promotions in MarketingDocument121 pagesPromotions in MarketingRishabh Jain100% (1)
- Angostura - Vals Venezolano Tab by Antonio Lauro at Ultimate-GuitarDocument3 pagesAngostura - Vals Venezolano Tab by Antonio Lauro at Ultimate-GuitarHarold CamayaguanNo ratings yet
- Iso 7500-1-2015Document24 pagesIso 7500-1-2015Иван ИвановNo ratings yet
- Listening Test 1Document4 pagesListening Test 1estefania aranda jimenezNo ratings yet
- Dune 2000 Gruntmods Edition ManualDocument43 pagesDune 2000 Gruntmods Edition Manualkrishna murtiNo ratings yet
- Aveva NETDocument8 pagesAveva NETchandru683No ratings yet
- Inventive Pricing of Urban Public TransportDocument19 pagesInventive Pricing of Urban Public TransportSean100% (1)
- 2 - Airframes & SystemsDocument36 pages2 - Airframes & SystemsJoe ThompsonNo ratings yet
- Algorithms - The University of SydneyDocument6 pagesAlgorithms - The University of SydneySuman.SNo ratings yet
- International Business DirectoryDocument1 pageInternational Business DirectoryIvy Michelle HabagatNo ratings yet
- Module 2 NSTP 1 Values and Ethics 1Document14 pagesModule 2 NSTP 1 Values and Ethics 1Master HaroldNo ratings yet
- V-2154-101-A-111 - 0A Method Statement Stairway Tank InstallationDocument24 pagesV-2154-101-A-111 - 0A Method Statement Stairway Tank InstallationWidya PrasetyaNo ratings yet
- 12.Dr. Sheela Srivastava Mr. M. Sivakoti Reddy Vol.2, Issue 1 & 2 PDFDocument3 pages12.Dr. Sheela Srivastava Mr. M. Sivakoti Reddy Vol.2, Issue 1 & 2 PDFMuhammad Ibrahim100% (1)
- E-Auctions - MSTC Limited-DVC-MEJIA POWER-07.08.2019Document10 pagesE-Auctions - MSTC Limited-DVC-MEJIA POWER-07.08.2019mannakauNo ratings yet
- Forward ConverterDocument31 pagesForward ConverterHitarth BuchNo ratings yet
- Law Landmark CasesDocument107 pagesLaw Landmark CasesjayNo ratings yet
- BSBLDR401 Task2Document13 pagesBSBLDR401 Task2Winnie PPnapatNo ratings yet