You might also like
- 4 Obstructive UropathyDocument37 pages4 Obstructive UropathyCoy NuñezNo ratings yet
- Charts For Kidney and Lower Urinary Tract Pathology. NephrologyDocument34 pagesCharts For Kidney and Lower Urinary Tract Pathology. NephrologyM PatelNo ratings yet
- Scanning Technique of KidneysDocument103 pagesScanning Technique of KidneysPhuntsho OngmoNo ratings yet
- Renal Tumors: By: DR Haseeb KhanDocument52 pagesRenal Tumors: By: DR Haseeb KhanammaraNo ratings yet
- Plant Tissue Culture (Pharmacognosy)Document31 pagesPlant Tissue Culture (Pharmacognosy)anas ab100% (5)
- Newborn ExaminationDocument45 pagesNewborn ExaminationHamka HamNo ratings yet
- Toxic Tooth Root Canal Mercola InterviewDocument13 pagesToxic Tooth Root Canal Mercola InterviewAntonio PitaguariNo ratings yet
- BLOOD - REVIEWERDocument6 pagesBLOOD - REVIEWERVanya YeleniaNo ratings yet
- 09 - Congenital SyndromesDocument76 pages09 - Congenital SyndromesROHIT100% (1)
- Mnlkaxi QH: Manisha M.Sc. NursingDocument57 pagesMnlkaxi QH: Manisha M.Sc. NursingManisha ShakyaNo ratings yet
- Nephrotic Syndrome What Is Nephrotic Syndrome? CauseDocument10 pagesNephrotic Syndrome What Is Nephrotic Syndrome? CauseThuganamix50% (2)
- Nada's GI Path ReviewDocument44 pagesNada's GI Path ReviewNada Much100% (2)
- Water Carbon Nitrogen: Presented by Aditya KumarDocument16 pagesWater Carbon Nitrogen: Presented by Aditya KumarAditya kumarNo ratings yet
- DR Mansi Thesis ProtocolDocument13 pagesDR Mansi Thesis ProtocolAditya kumarNo ratings yet
- Kawasaki DiseaseDocument41 pagesKawasaki DiseaseWuryan Dewi Mftahtyas ArumNo ratings yet
- CASE PRE - Acute Lymphoblastic LeukemiaDocument153 pagesCASE PRE - Acute Lymphoblastic Leukemiacecille_javier100% (5)
- Turner Syndrome, Klinefelter Syndrome, Down SyndromeDocument78 pagesTurner Syndrome, Klinefelter Syndrome, Down SyndromeTasya100% (3)
- The Slim Book of Health Pearls: Symptoms Never to IgnoreFrom EverandThe Slim Book of Health Pearls: Symptoms Never to IgnoreNo ratings yet
- MCQS On Protein Metabolism BY Siraj Ul IslamDocument12 pagesMCQS On Protein Metabolism BY Siraj Ul Islamsirajkhan93100% (2)
- Congenital Anomalies of KidneDocument7 pagesCongenital Anomalies of KidneSanthosh.S.U100% (2)
- Rare DiseasesDocument15 pagesRare DiseasesTaimur Khalil100% (1)
- Early Diagnosis of MalignancyDocument29 pagesEarly Diagnosis of Malignancyokwadha simionNo ratings yet
- NeonatologyDocument42 pagesNeonatologybolt boltNo ratings yet
- 9.emma Renal Cysts Hanoi 2018Document39 pages9.emma Renal Cysts Hanoi 2018vd hungNo ratings yet
- Diseases of Urinary SystemDocument29 pagesDiseases of Urinary SystemHassan.shehri100% (9)
- Oesphageal Cancer: DR Ka NaiduDocument48 pagesOesphageal Cancer: DR Ka NaiduKISHAN NAIDUNo ratings yet
- Legg Calvé Perthes Disease 1Document66 pagesLegg Calvé Perthes Disease 1Vignesh WaranNo ratings yet
- Fabry DiseaseDocument63 pagesFabry DiseaseKunal PaulNo ratings yet
- 2 Nasopharyngeal Dise (Sept. 2019) - StudentDocument80 pages2 Nasopharyngeal Dise (Sept. 2019) - StudentNicNo ratings yet
- WILMS TUMOR NewDocument22 pagesWILMS TUMOR NewBSN2G- SABLA-ON LORRAINE ANNENo ratings yet
- Case Presentation: Baguio General Hospital and Medical Center Department of General SurgeryDocument27 pagesCase Presentation: Baguio General Hospital and Medical Center Department of General SurgeryJorge De VeraNo ratings yet
- Basal Cell Carcinoma of The Eye Lid by Thea Umbac, Ophthalmology InternDocument32 pagesBasal Cell Carcinoma of The Eye Lid by Thea Umbac, Ophthalmology InternThea SelinaNo ratings yet
- Head and Neck Cancers-Predisposing Factors-Etiology-Premalign LesionsDocument41 pagesHead and Neck Cancers-Predisposing Factors-Etiology-Premalign Lesionspianistgirl99No ratings yet
- Deteksi Dini Kelainan Pada JaninDocument108 pagesDeteksi Dini Kelainan Pada JaninYuyunNo ratings yet
- Pediatric Head and Neck Tumors: Hisham Dahmoush, MBBCH FRCR Stanford RadiologyDocument78 pagesPediatric Head and Neck Tumors: Hisham Dahmoush, MBBCH FRCR Stanford RadiologykathyNo ratings yet
- ADPKDDocument75 pagesADPKDVenkataramanan KrishnamoorthyNo ratings yet
- Kidney & Urinary Tract USDocument62 pagesKidney & Urinary Tract USBayarbaatar BoldNo ratings yet
- Deep Vein Thrombosis: Carla C. Fernández Santos Universidad Central Del Caribe, Bayamón, PR Family MedicineDocument20 pagesDeep Vein Thrombosis: Carla C. Fernández Santos Universidad Central Del Caribe, Bayamón, PR Family MedicineCarla C. FernandezNo ratings yet
- PediaDocument10 pagesPediaKannan KannanNo ratings yet
- Hydrocephalus: Lambertus Josef F. BugaDocument25 pagesHydrocephalus: Lambertus Josef F. BugaPraktek Dokter Umum drjosefdrelsyNo ratings yet
- Blood DisordersDocument96 pagesBlood DisordersIndragee EkanayakeNo ratings yet
- Urology (CTIN) V2Document45 pagesUrology (CTIN) V2Fathy ElsheshtawyNo ratings yet
- Pediatric Puzzler: October 30, 2007 Rachel and Caroline, Mds Best Peds Chiefs EverDocument37 pagesPediatric Puzzler: October 30, 2007 Rachel and Caroline, Mds Best Peds Chiefs EverTaufan LutfiNo ratings yet
- CAKUTDocument50 pagesCAKUTsantosh subediNo ratings yet
- Prepared By: Dr. Abhishek Garg M.D Resident 3 Year Edited By: Dr. Arun Kumar SharmaDocument43 pagesPrepared By: Dr. Abhishek Garg M.D Resident 3 Year Edited By: Dr. Arun Kumar SharmaVijitha LenkalaNo ratings yet
- CPC Medical Oncology 2Document52 pagesCPC Medical Oncology 2Ravi KumarNo ratings yet
- Acute Appendic ItsDocument24 pagesAcute Appendic ItsDr-Wisam Mhmd AliNo ratings yet
- Kidney, Bladder and Prostate Pathology For Allied Health SciencesDocument38 pagesKidney, Bladder and Prostate Pathology For Allied Health SciencesMichael BrownNo ratings yet
- Wilson's Disease: Dr. Imtiyaz Hashim PGR Mu-Iv (B)Document14 pagesWilson's Disease: Dr. Imtiyaz Hashim PGR Mu-Iv (B)امتیاز ہاشم بزنجوNo ratings yet
- Turners Syndrome: Sonika Shimon Prasad S160232Document20 pagesTurners Syndrome: Sonika Shimon Prasad S160232Ivy DanNo ratings yet
- Week 7 Oncology and ChemotherapyDocument42 pagesWeek 7 Oncology and ChemotherapyJAN MARIE EVANGELISTANo ratings yet
- Polycystic Kidney by SantoshDocument32 pagesPolycystic Kidney by SantoshSantosh KumarNo ratings yet
- Test 2 NotesDocument38 pagesTest 2 Notesbjpalmer100% (2)
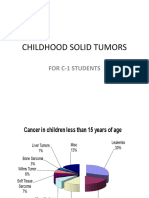
- Childhood Solid Tumors: For C-1 StudentsDocument72 pagesChildhood Solid Tumors: For C-1 StudentsYemata HailuNo ratings yet
- Patology Kidney: Dr. Delyuzar SP - PADocument71 pagesPatology Kidney: Dr. Delyuzar SP - PAJefry SNo ratings yet
- Oncology StarsDocument61 pagesOncology StarsCharles Conrad Pagcaliwagan BagsitNo ratings yet
- General Survey by DR ADBDocument34 pagesGeneral Survey by DR ADBRahul KumarNo ratings yet
- 07 - Basics of Adrenal Urinary Tract Prostate and Testicle ImagingDocument139 pages07 - Basics of Adrenal Urinary Tract Prostate and Testicle ImagingHusam AbuodehNo ratings yet
- History and Examination of Urologenital SystemDocument29 pagesHistory and Examination of Urologenital SystemPrincewill SeiyefaNo ratings yet
- General Physical Examination (GPE) in Neurology DR GaneshgoudaDocument120 pagesGeneral Physical Examination (GPE) in Neurology DR GaneshgoudaDr Ganeshgouda MajigoudraNo ratings yet
- Patho FinaaaaalllDocument15 pagesPatho FinaaaaalllIvin SajithNo ratings yet
- Achondroplasia: Disproportionate Short StatureDocument67 pagesAchondroplasia: Disproportionate Short StaturenuraswadNo ratings yet
- Epithelial Tumours - SreejaDocument184 pagesEpithelial Tumours - SreejaaakiNo ratings yet
- Annalyn S. Da-Anoy, M.D., R.M.T.Document88 pagesAnnalyn S. Da-Anoy, M.D., R.M.T.api-25914483No ratings yet
- Alport'S SyndromeDocument8 pagesAlport'S SyndromeHemanth PrakashNo ratings yet
- Hepatosplenomegaly: Dr. C.V. RavisekarDocument16 pagesHepatosplenomegaly: Dr. C.V. RavisekarnilmbbsNo ratings yet
- Physics InvestigatoryDocument9 pagesPhysics InvestigatoryAditya kumarNo ratings yet
- Practice Test Paper For Class 9 English 2017-2018Document12 pagesPractice Test Paper For Class 9 English 2017-2018Aditya kumarNo ratings yet
- Aditya Kumar Chemistry ProjectDocument15 pagesAditya Kumar Chemistry ProjectAditya kumar0% (1)
- Physical Education Class 11 Long Answers Chapter 4Document5 pagesPhysical Education Class 11 Long Answers Chapter 4Aditya kumarNo ratings yet
- Cbse Question Paper Class-X: Mathematics I1fu1aDocument15 pagesCbse Question Paper Class-X: Mathematics I1fu1aAditya kumarNo ratings yet
- Gentics PDFDocument7 pagesGentics PDFHarpreet KaurNo ratings yet
- Laboratory Technology Course Catalogue LastDocument9 pagesLaboratory Technology Course Catalogue LastBiruk MilionNo ratings yet
- Chemo Practice ProblemsDocument12 pagesChemo Practice ProblemsSamNo ratings yet
- Hema PrescriptionDocument56 pagesHema PrescriptionLeslie Noelle GarciaNo ratings yet
- Virological and Immunological Effects of AntioxidantDocument11 pagesVirological and Immunological Effects of AntioxidantHector Javier Chavez RamirezNo ratings yet
- Haematology PhysiologyDocument15 pagesHaematology PhysiologyOlivia LimNo ratings yet
- Origin of Replication PDFDocument9 pagesOrigin of Replication PDFinoka911No ratings yet
- Chapter 1 Nerve Cells and Nerve Impulses 2Document19 pagesChapter 1 Nerve Cells and Nerve Impulses 2REPALDA, Irish N.No ratings yet
- 7mm Frog Embryo - EMBLabDocument3 pages7mm Frog Embryo - EMBLabIvy Cruz100% (1)
- Sickle Cell Student-LabDocument12 pagesSickle Cell Student-LabS. Spencer100% (1)
- Title: To Investigate The Effectiveness of Antibiotics in Inhibiting Growth of Bacterial CellDocument13 pagesTitle: To Investigate The Effectiveness of Antibiotics in Inhibiting Growth of Bacterial CellGayathri GunasekaranNo ratings yet
- GM CropsDocument2 pagesGM CropsLavanya DevulapalliNo ratings yet
- How Human Medicine Could Save The Tasmanian DevilDocument4 pagesHow Human Medicine Could Save The Tasmanian DevilGiang NguyenNo ratings yet
- KFN 040Document11 pagesKFN 040Larissa SavadogoNo ratings yet
- 38 - Epithelial Attachment and Downgrowth On Dental ImplantDocument9 pages38 - Epithelial Attachment and Downgrowth On Dental ImplantPablo BenitezNo ratings yet
- Endorsement Letter - July 4, 2018Document3 pagesEndorsement Letter - July 4, 2018Ángela BernardoNo ratings yet
- Mind As Epigenetic Modifier in Mood DisordersDocument7 pagesMind As Epigenetic Modifier in Mood DisordersSabra PelhamNo ratings yet
- Thyrocare Reat Liest 2023Document6 pagesThyrocare Reat Liest 2023dineshNo ratings yet
- Dr. Asifa Iqbal Oral and Maxillofacial Surgery, KEMU Mayo Hospital, LahoreDocument53 pagesDr. Asifa Iqbal Oral and Maxillofacial Surgery, KEMU Mayo Hospital, LahoreAl RawdhaNo ratings yet
- Thygeson's Superficial Punctate Keratitis: Signs and SymptomsDocument3 pagesThygeson's Superficial Punctate Keratitis: Signs and SymptomsYulia KasihNo ratings yet
- Celts Descended From SpainDocument2 pagesCelts Descended From SpainTHX55No ratings yet
- Dietary Intervention in AcneDocument13 pagesDietary Intervention in AcnePhelipe Auerswald Do AmaralNo ratings yet
- Kuliah Farmakologi - Farmakokinetik Dan Farmakodinamik Antivirus Dan Antiparasit Secara UmumDocument87 pagesKuliah Farmakologi - Farmakokinetik Dan Farmakodinamik Antivirus Dan Antiparasit Secara UmumGabriel AnindhitaNo ratings yet
- Phycocyanin A Potential Drug For Cancer TreatmentDocument14 pagesPhycocyanin A Potential Drug For Cancer TreatmentSrivatsava RajagopalanNo ratings yet
- Hip Dysplasia in Dogs: A Guide For Dog OwnersDocument6 pagesHip Dysplasia in Dogs: A Guide For Dog Ownerstaner_soysurenNo ratings yet