You might also like
- The Vanishing Present: Wisconsin's Changing Lands, Waters, and WildlifeFrom EverandThe Vanishing Present: Wisconsin's Changing Lands, Waters, and WildlifeDonald M. WallerRating: 4 out of 5 stars4/5 (1)
- Tbi Tasa Pro 2004-2016 Prov NombresDocument1 pageTbi Tasa Pro 2004-2016 Prov NombresRebeca Bravo PradoNo ratings yet
- The Rough Guide to Belize (Travel Guide eBook): with Tikal and FloresFrom EverandThe Rough Guide to Belize (Travel Guide eBook): with Tikal and FloresNo ratings yet
- Major RORO Routes MapDocument1 pageMajor RORO Routes MapRolando Tacneng QuerubinNo ratings yet
- Safety Nets in Africa: Effective Mechanisms to Reach the Poor and Most VulnerableFrom EverandSafety Nets in Africa: Effective Mechanisms to Reach the Poor and Most VulnerableNo ratings yet
- Vicinity Map LinaboDocument1 pageVicinity Map LinaboJunar PlagaNo ratings yet
- Road Map BasilanDocument1 pageRoad Map BasilanVeronica AlvaradoNo ratings yet
- Roulet eDocument1 pageRoulet eJan JanNo ratings yet
- Magsaysay: La Beb Gen. MdseDocument1 pageMagsaysay: La Beb Gen. MdseAhna Manahan MercurioNo ratings yet
- This Site: River Control Typical Cross SectionDocument1 pageThis Site: River Control Typical Cross SectionjohnNo ratings yet
- Región de La AraucaníaDocument11 pagesRegión de La AraucaníaCristian NeiraNo ratings yet
- METRADO AGUA POTABLE LA JOYA FINAL (F)Document4 pagesMETRADO AGUA POTABLE LA JOYA FINAL (F)Yarid Colonio ayquipaNo ratings yet
- StarMap October 2017Document1 pageStarMap October 2017TibzNo ratings yet
- Allergens AUG23Document22 pagesAllergens AUG23Rogério LimaNo ratings yet
- Camp Calendars - Camp KaylieDocument1 pageCamp Calendars - Camp KayliejcjjcjdkdNo ratings yet
- Ruv-Ruben Mejia 2023Document2 pagesRuv-Ruben Mejia 2023alexNo ratings yet
- Catalogo Alimentacion GULFOOD 2019 DUBAIDocument72 pagesCatalogo Alimentacion GULFOOD 2019 DUBAIJordi MaciasNo ratings yet
- 2010 Schedule With PromotionsDocument1 page2010 Schedule With PromotionsCarissa NicholsNo ratings yet
- Downloaded From Https://read - Dukeupress.edu/books/chapter-Pdf/515475/9780822373865-001.pdf by University of Michigan User On 13 February 2018Document9 pagesDownloaded From Https://read - Dukeupress.edu/books/chapter-Pdf/515475/9780822373865-001.pdf by University of Michigan User On 13 February 2018PabloNo ratings yet
- Control de Avance - Alto y Bajo 30-01-21Document5 pagesControl de Avance - Alto y Bajo 30-01-21KATHIA MILAGRITOS SANCHEZ CIPRIANONo ratings yet
- Philippine Statistics Authority, 2017 Top Producer: RegionsDocument2 pagesPhilippine Statistics Authority, 2017 Top Producer: RegionsJayson BasiagNo ratings yet
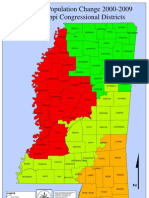
- Mississippi Population Changes by Congressional District (00-09)Document2 pagesMississippi Population Changes by Congressional District (00-09)Majority In Mississippi blogNo ratings yet
- Nile Delta Strat. Col.Document1 pageNile Delta Strat. Col.Mohamed Abd El-ma'boudNo ratings yet
- Peta Taliabu Untuk Avensa MapDocument1 pagePeta Taliabu Untuk Avensa MapNonk Moloku Mbah VulcanoNo ratings yet
- Presenters: Jose Rojo G. Alisla Rosemarie S. GumeraDocument26 pagesPresenters: Jose Rojo G. Alisla Rosemarie S. GumeraĐương Anh CaoNo ratings yet
- 2016 TotaalDocument3 pages2016 TotaalCarlos AlanisNo ratings yet
- b2207b 508Document60 pagesb2207b 508SynNo ratings yet
- LISTODocument35 pagesLISTOYang Rhea100% (2)
- Garapua KeyfactsDocument8 pagesGarapua KeyfactsgmigukNo ratings yet
- Printable Intramuros MapDocument6 pagesPrintable Intramuros MapAe R ON100% (1)
- Culinary Schedule Abril 2022 Del 4 Al 10 de AbrilDocument2 pagesCulinary Schedule Abril 2022 Del 4 Al 10 de AbrilErick PedrazaNo ratings yet
- Development - Economics by Dwight PerkinsDocument875 pagesDevelopment - Economics by Dwight PerkinsTECH TUBENo ratings yet
- Vicinity MapDocument1 pageVicinity MapLimwell AquinoNo ratings yet
- Bukaan 23 September 2023 (September-59)Document5 pagesBukaan 23 September 2023 (September-59)sinta anggrainiNo ratings yet
- Sep AklanDocument235 pagesSep Aklanapi-3855623100% (2)
- BCMC SangayDocument3 pagesBCMC SangayJet JetNo ratings yet
- MAPA BASE DE CASCAVEL CENTRO - Layout2Document1 pageMAPA BASE DE CASCAVEL CENTRO - Layout2Roberto ZanonNo ratings yet
- Il CoDocument1 pageIl CoAndrew SobolNo ratings yet
- Location Plan: M Ap of MindanaoDocument1 pageLocation Plan: M Ap of MindanaoJunar PlagaNo ratings yet
- You Created This PDF From An Application That Is Not Licensed To Print To Novapdf PrinterDocument1 pageYou Created This PDF From An Application That Is Not Licensed To Print To Novapdf PrinterRond YanNo ratings yet
- The Deep Sea: The Twilight Zone The Twilight ZoneDocument1 pageThe Deep Sea: The Twilight Zone The Twilight ZoneSammy WilliamsNo ratings yet
- Mapa Coacalco A SulzerDocument3 pagesMapa Coacalco A SulzerGabino TorresNo ratings yet
- MAPA BASE DE CASCAVEL NOVO-Layout2Document1 pageMAPA BASE DE CASCAVEL NOVO-Layout2Roberto ZanonNo ratings yet
- A Dónde Querés Ir - AbastoDocument4 pagesA Dónde Querés Ir - AbastomathevsfpNo ratings yet
- Spectrum-Assignments 202012 2.3GHzDocument1 pageSpectrum-Assignments 202012 2.3GHzOje Abdul-QuadriNo ratings yet
- Wine Yeast ChartDocument1 pageWine Yeast ChartDan Mihai RusuNo ratings yet
- Stratigraphy Column Ikpikpuk 1: Chrono - Stratigraphy Lithostratigrafhy Lithology Sismkco Profile Hydrocarbon PotencialDocument2 pagesStratigraphy Column Ikpikpuk 1: Chrono - Stratigraphy Lithostratigrafhy Lithology Sismkco Profile Hydrocarbon PotencialJose Cartagena ZubietaNo ratings yet
- Sportsmansguidet 00 AitkDocument62 pagesSportsmansguidet 00 Aitkemerson.kafkaNo ratings yet
- Keyboard Er159Document1 pageKeyboard Er159John sNo ratings yet
- Anderson 2020Document160 pagesAnderson 2020LuisFer CastamanNo ratings yet
- 4id Security Situation Update For Joint RDC-RPOC 10 Mar 2Document20 pages4id Security Situation Update For Joint RDC-RPOC 10 Mar 2Enp Titus VelezNo ratings yet
- HW 01 220Document3 pagesHW 01 220zhang JayantNo ratings yet
- Distribution of PopulationDocument1 pageDistribution of Populationapi-3701421100% (3)
- MUH051069 Star Trek Adventures - Delta Quadrant (Printer Friendly) (OEF) (2020) PDFDocument144 pagesMUH051069 Star Trek Adventures - Delta Quadrant (Printer Friendly) (OEF) (2020) PDFsickbastard2391% (11)
- 2017-2019 Pops Plan FinalDocument39 pages2017-2019 Pops Plan FinalByaheng CawayanNo ratings yet
- Mapa Del Peru: Ecuador ColombiaDocument1 pageMapa Del Peru: Ecuador Colombiayon yonatan tintaya capquequiNo ratings yet
- Elevation Map of The Northern Territory: Timor SEA Arafura SeaDocument1 pageElevation Map of The Northern Territory: Timor SEA Arafura SeaamooooosNo ratings yet
- Mapa Politico de NriñoDocument1 pageMapa Politico de NriñoYedison Fernando Prieto PrietoNo ratings yet
- San Nicolas: Bulacan Province Bulacan Province SmiaDocument1 pageSan Nicolas: Bulacan Province Bulacan Province SmiaEric LaluanNo ratings yet
- Grupo Cuyo en La Dorsal de Huincul (Gómez Omil Et Al, 2002)Document22 pagesGrupo Cuyo en La Dorsal de Huincul (Gómez Omil Et Al, 2002)Ariel MichelettoNo ratings yet
- Respirology Adult Sample Polysomnography Report eDocument2 pagesRespirology Adult Sample Polysomnography Report eignatiaratna prativiNo ratings yet
- Cerebrovascular AccidentDocument10 pagesCerebrovascular Accidentcarls burg a. resurreccionNo ratings yet
- Coek - Info - Neurocomputing Foundations of ResearchDocument5 pagesCoek - Info - Neurocomputing Foundations of Researchkgdeepak122950No ratings yet
- Medical Hypotheses: Gary Steinman, David Mankuta TDocument3 pagesMedical Hypotheses: Gary Steinman, David Mankuta TveronicaNo ratings yet
- EnhancingDocument17 pagesEnhancingJosephKiwasLlamido100% (1)
- Patrick Colm Hogan Affective Narratology. The Emotional Structure of StoriesDocument305 pagesPatrick Colm Hogan Affective Narratology. The Emotional Structure of StoriesNathan Wagar100% (4)
- Bodynamic Character Analysis GuideDocument22 pagesBodynamic Character Analysis GuideClara Marina Galliez100% (1)
- How The Brain Experiences ArchitectureDocument16 pagesHow The Brain Experiences ArchitectureAbdu Thora100% (1)
- General Psychology Chapter 4Document3 pagesGeneral Psychology Chapter 4redina021No ratings yet
- Reading SkillsDocument4 pagesReading SkillsAmina Ait SaiNo ratings yet
- Theories of Child and Adolescent DevelopmentDocument48 pagesTheories of Child and Adolescent DevelopmentJela Marie CandaNo ratings yet
- 02 Fundamentals of Behavioral Neurology - CohenDocument24 pages02 Fundamentals of Behavioral Neurology - CohenSarah SabtiNo ratings yet
- Portfolio Lola DiazDocument7 pagesPortfolio Lola DiazLola Díaz PrietoNo ratings yet
- Clinicians Club The Anxiety and Depression Workbook Supplemental Guide For CliniciansDocument50 pagesClinicians Club The Anxiety and Depression Workbook Supplemental Guide For CliniciansJuan Alberto González100% (2)
- Robert Dilts - Eye Scanning PatternsDocument5 pagesRobert Dilts - Eye Scanning PatternsGarb PoubNo ratings yet
- Social Psychology: Lecture 7.1: Is Empathy A Magic Bullet? The Power of OutrospectionDocument3 pagesSocial Psychology: Lecture 7.1: Is Empathy A Magic Bullet? The Power of OutrospectionchandanNo ratings yet
- Reflections (Remembering and Forgetting)Document1 pageReflections (Remembering and Forgetting)ShaniceNo ratings yet
- Preclinical Evaluation of Anti-Epileptics: S K Kanthlal Dept of Pharmacology Amrita School of Pharmacy KochiDocument24 pagesPreclinical Evaluation of Anti-Epileptics: S K Kanthlal Dept of Pharmacology Amrita School of Pharmacy Kochigunuputi sushmaNo ratings yet
- Childhood, Growing Up & Learning (BSCBED-151) IASE SardarsaharDocument4 pagesChildhood, Growing Up & Learning (BSCBED-151) IASE Sardarsaharhem rajNo ratings yet
- Advertising and The Seven Sins of MemoryDocument6 pagesAdvertising and The Seven Sins of MemoryRahul Pachori100% (1)
- Learning TargetsDocument1 pageLearning TargetsCarlos Leo SantosNo ratings yet
- ACT Guide FinalDocument25 pagesACT Guide Finalrama krishna100% (3)
- Neuroscience of Addiction Review: George F. Koob, Pietro Paolo Sanna, and Floyd E. BloomDocument10 pagesNeuroscience of Addiction Review: George F. Koob, Pietro Paolo Sanna, and Floyd E. Bloomadri90No ratings yet
- TMP E946Document7 pagesTMP E946FrontiersNo ratings yet
- Science of Psychology An Appreciative View 4Th Edition King Solutions Manual Full Chapter PDFDocument57 pagesScience of Psychology An Appreciative View 4Th Edition King Solutions Manual Full Chapter PDFgenericparaxialb41vmq100% (9)
- Actual4Test: Actual4test - Actual Test Exam Dumps-Pass For IT ExamsDocument4 pagesActual4Test: Actual4test - Actual Test Exam Dumps-Pass For IT ExamsBryan OrdoñezNo ratings yet
- A Teacher Used The Materials Like TVDocument2 pagesA Teacher Used The Materials Like TVEva NolascoNo ratings yet
- Behavioral Learning TheoryDocument26 pagesBehavioral Learning TheoryAda Gay Olandia Serencio50% (2)
- Project Report Role of Emotional Inntelligence in Employee Performance (COLLEGE)Document46 pagesProject Report Role of Emotional Inntelligence in Employee Performance (COLLEGE)Sankalp GahlotNo ratings yet
- Book Review An Action Plan For Your Inner Child Parenting Each OtherDocument3 pagesBook Review An Action Plan For Your Inner Child Parenting Each OtherNarcis NagyNo ratings yet