You might also like
- Chemical Kinetics: - : Types of Chemical ReactionsDocument12 pagesChemical Kinetics: - : Types of Chemical ReactionsmanishNo ratings yet
- Chemical Kinetics Neet MCQDocument12 pagesChemical Kinetics Neet MCQmanan10jas1529No ratings yet
- Chemical KineticsDocument72 pagesChemical KineticsSiddhartha KumarNo ratings yet
- Identifikasi KationDocument106 pagesIdentifikasi KationHAIDAR RACHMANNo ratings yet
- Kinetics UkgvdWVDocument17 pagesKinetics UkgvdWVMaica GarampilNo ratings yet
- Chemical KineticsDocument21 pagesChemical Kineticsdipankargh48No ratings yet
- 22 00 14 11 12 2023 Doc-20220901-Wa0007.Document9 pages22 00 14 11 12 2023 Doc-20220901-Wa0007.hmegm123No ratings yet
- Chemical Kinetics REVISEDocument62 pagesChemical Kinetics REVISEpriyapriyankan43No ratings yet
- CH 2. Chemical Kinetics (Chem +2)Document55 pagesCH 2. Chemical Kinetics (Chem +2)Yash RotteNo ratings yet
- Chemical Kinetics: Rate of A ReactionDocument49 pagesChemical Kinetics: Rate of A ReactionVijay KumarNo ratings yet
- 2) 2020 - Chemical - KineticsDocument8 pages2) 2020 - Chemical - KineticsFaizan AnsariNo ratings yet
- Chemical1.2lecture SlidesDocument128 pagesChemical1.2lecture SlidesMUHAMMAD SHAHBAZNo ratings yet
- Chemical KineticsDocument49 pagesChemical KineticsS KNo ratings yet
- Kinetics OverviewDocument70 pagesKinetics Overviewdsagnik72No ratings yet
- Chemical KineticsDocument102 pagesChemical KineticsKishore SurampalliNo ratings yet
- Chemical Kinetics: T 0 ' o D (R) D (P) R DT DTDocument12 pagesChemical Kinetics: T 0 ' o D (R) D (P) R DT DTVeerNo ratings yet
- KineticsDocument37 pagesKineticsJessika DorssersNo ratings yet
- Chemical KineticsDocument64 pagesChemical KineticsFerdiansyah SetiawanNo ratings yet
- Chemical Kinetics/Rate of Chemical ReactionDocument13 pagesChemical Kinetics/Rate of Chemical ReactionHiNo ratings yet
- Che 156 - Chemical Kinetics Unit 1-6 MergedDocument165 pagesChe 156 - Chemical Kinetics Unit 1-6 MergedIsrael OluwayomiNo ratings yet
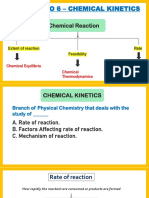
- Chapter No 6 - Chemical KineticsDocument45 pagesChapter No 6 - Chemical KineticsTanish SalviNo ratings yet
- Chemical Kinetics - Reaction RatesDocument35 pagesChemical Kinetics - Reaction RatesRichie SuyaoNo ratings yet
- C1 Reaction KineticsDocument12 pagesC1 Reaction KineticsChloeNo ratings yet
- Kinetics (Filled)Document36 pagesKinetics (Filled)MarikNo ratings yet
- Chemical Kinetics Lecture 1 2Document59 pagesChemical Kinetics Lecture 1 2BaNcHoNo ratings yet
- Topic 1 and 2-ChemicalKineticsDocument86 pagesTopic 1 and 2-ChemicalKineticsNOR AZAM BIN ENDOT / FSNo ratings yet
- Chemical Kinetics & Reactor DesignDocument134 pagesChemical Kinetics & Reactor DesignShakoor MalikNo ratings yet
- Chemical KineticsDocument32 pagesChemical KineticsTimothy HandokoNo ratings yet
- Chemical KineticDocument40 pagesChemical KineticHamzaNo ratings yet
- Chemical KineticsDocument39 pagesChemical Kineticsneel-amberNo ratings yet
- All Combine Chemical Eq28.02.23Document27 pagesAll Combine Chemical Eq28.02.23kaustabh2005No ratings yet
- Chemical KineticsDocument101 pagesChemical Kineticsec1412No ratings yet
- 01-Chep-11-Chemical Kinetics-Theory-Final-EDocument8 pages01-Chep-11-Chemical Kinetics-Theory-Final-EAbhishek RavirajNo ratings yet
- Chemical KineticsDocument9 pagesChemical KineticsMikey Bryant BonbonNo ratings yet
- Chemical and Enzyme Kinetics Lecture 2Document47 pagesChemical and Enzyme Kinetics Lecture 2downdstairs45No ratings yet
- Chemicalkinetics Presentation 150214034801 Conversion Gate02Document35 pagesChemicalkinetics Presentation 150214034801 Conversion Gate02BLACK HACKERNo ratings yet
- Kinetics OverviewDocument88 pagesKinetics OverviewMovie LitNo ratings yet
- CHM131 - Chapter 7 - Chemical EquilibriumDocument30 pagesCHM131 - Chapter 7 - Chemical EquilibriumNotes NotesNo ratings yet
- Chapter 14 PDF File of Power PointDocument78 pagesChapter 14 PDF File of Power PointNair CastilloNo ratings yet
- Kinetics LPDocument41 pagesKinetics LPHarkritSinghNo ratings yet
- Lecture 4 - Chem Kinetics - Modified GutoDocument40 pagesLecture 4 - Chem Kinetics - Modified GutoSamwel MwetichNo ratings yet
- Chemical Kinetics - Reaction Orders: H° For This Reaction Is: H H HDocument13 pagesChemical Kinetics - Reaction Orders: H° For This Reaction Is: H H HLinaGeneyReyesNo ratings yet
- Kinetics OverviewDocument44 pagesKinetics OverviewLogesh BalamuruganNo ratings yet
- Chemical Kinetics and ColloidsDocument6 pagesChemical Kinetics and Colloidstahasheikh822No ratings yet
- Chemical KineticsDocument15 pagesChemical KineticsThara BijuNo ratings yet
- Reactor Technology 6Document13 pagesReactor Technology 6Sami WhiteNo ratings yet
- Essentials of Chemical KineticsDocument49 pagesEssentials of Chemical KineticsJohn KanteNo ratings yet
- Chemical KineticsDocument56 pagesChemical KineticsMohamed KhaledNo ratings yet
- Chapter 4Document32 pagesChapter 4Nguyen NhatNo ratings yet
- Chemical KineticsDocument3 pagesChemical KineticsRachel AustriaNo ratings yet
- Chemical KineticsDocument32 pagesChemical KineticsRaju SinghNo ratings yet
- Kinetics IntroductionDocument55 pagesKinetics IntroductionAgano juma mwakasendoNo ratings yet
- Shailendra KR.: Physical ChemistryDocument13 pagesShailendra KR.: Physical ChemistryAli SaabNo ratings yet
- 7 Reaction KineticsDocument38 pages7 Reaction KineticsArvin LiangdyNo ratings yet
- Chemical KineticsDocument77 pagesChemical KineticsDipu RokayaNo ratings yet
- A Modern Course in Statistical PhysicsFrom EverandA Modern Course in Statistical PhysicsRating: 3.5 out of 5 stars3.5/5 (2)
- Critical Evaluation of Equilibrium Constants Involving 8-Hydroxyquinoline and Its Metal Chelates: Critical Evaluation of Equilibrium Constants in Solution: Part B: Equilibrium Constants of Liquid-Liquid Distribution SystemsFrom EverandCritical Evaluation of Equilibrium Constants Involving 8-Hydroxyquinoline and Its Metal Chelates: Critical Evaluation of Equilibrium Constants in Solution: Part B: Equilibrium Constants of Liquid-Liquid Distribution SystemsNo ratings yet
- Hardness Conversion ChartDocument3 pagesHardness Conversion ChartUlung SadewoNo ratings yet
- Tds-Bentone Hydroclay 550Document2 pagesTds-Bentone Hydroclay 550July CapilloNo ratings yet
- GeneralChemistry1 Q1 Mod4 Chemical-Formulas Ver-5Document21 pagesGeneralChemistry1 Q1 Mod4 Chemical-Formulas Ver-5JESSAMEN DOLORICAN100% (1)
- The Structure and Binding Mode of Citrate in The Stabilization of Gold NanoparticlesDocument6 pagesThe Structure and Binding Mode of Citrate in The Stabilization of Gold NanoparticlesRizkilukumNo ratings yet
- 2 5 23 Sialic Acid in Polysaccharide VaccinesDocument1 page2 5 23 Sialic Acid in Polysaccharide VaccinesLTV TM DVNo ratings yet
- 3 Chemistry End of Year Assessment Answer KeyDocument5 pages3 Chemistry End of Year Assessment Answer KeyMylene Heraga100% (1)
- HPLC Troubleshooting: Problem Area: PeaksDocument11 pagesHPLC Troubleshooting: Problem Area: PeaksMd. Ahedul IslamNo ratings yet
- A Combined Ion Exchange-Nanofiltration Process For Water Desalination - I. Chloride Sulphate Exchange PDFDocument7 pagesA Combined Ion Exchange-Nanofiltration Process For Water Desalination - I. Chloride Sulphate Exchange PDFalabdulgaderNo ratings yet
- ACFrOgDhnn MDm94k4jZjlj4SrU7oM5DNng3T7HppnXcS3i3ZIMzh5vTQOh11HQRzc5rTbD52UFYq BLIp2nfB4QJqYugseyksrsSZ UDYOFJg2fT1WAPPjjem6lWA0F 95gJmQ7lYf WuQs7fQfDocument3 pagesACFrOgDhnn MDm94k4jZjlj4SrU7oM5DNng3T7HppnXcS3i3ZIMzh5vTQOh11HQRzc5rTbD52UFYq BLIp2nfB4QJqYugseyksrsSZ UDYOFJg2fT1WAPPjjem6lWA0F 95gJmQ7lYf WuQs7fQfBiswanath BiswasNo ratings yet
- MOQ Project-Rev1Document17 pagesMOQ Project-Rev1Abhilash Kumar SinghNo ratings yet
- Class X - Summer ProjectsDocument6 pagesClass X - Summer ProjectsAsjad Ali KhanNo ratings yet
- ARTIGOCOBEMDocument11 pagesARTIGOCOBEMPradeep DonthaNo ratings yet
- Exp No-5Document15 pagesExp No-5shiamNo ratings yet
- Laboratory Result Proteolytic EnzymeDocument2 pagesLaboratory Result Proteolytic EnzymeWinter SnowNo ratings yet
- Cheese Lab ReportDocument4 pagesCheese Lab ReportJanelle-Janice Burgos0% (1)
- Synthesis and Reactivity in Inorganic and Metal-Organic ChemistryDocument18 pagesSynthesis and Reactivity in Inorganic and Metal-Organic ChemistryYoselin GomezNo ratings yet
- GB - T 14976-2012 Stainless Steel PipeDocument12 pagesGB - T 14976-2012 Stainless Steel PipeSK ChauNo ratings yet
- 2019f5s9ex4chemistry 1Document13 pages2019f5s9ex4chemistry 1Dania NatashaNo ratings yet
- Issues in Green ChemistryDocument8 pagesIssues in Green ChemistryYuxin XiaNo ratings yet
- Alternative Shaly Sands Water Saturation Equations ComparisonDocument32 pagesAlternative Shaly Sands Water Saturation Equations ComparisonKartiko WibowoNo ratings yet
- Experimental Studies On Utilization of Shredded Plastic Waste in Hardened Concrete MixDocument52 pagesExperimental Studies On Utilization of Shredded Plastic Waste in Hardened Concrete MixShashi KumarNo ratings yet
- Sample Questions - Chapter 13Document4 pagesSample Questions - Chapter 13Uday Prakash SahuNo ratings yet
- Astm D4060-10Document5 pagesAstm D4060-10Diomer Alzate Berrio100% (1)
- Cablemate Surface DuctingDocument6 pagesCablemate Surface Ductings11174444No ratings yet
- Novel Condensation of d-LA Into D-LSD Via PyBOPDocument1 pageNovel Condensation of d-LA Into D-LSD Via PyBOPX100% (1)
- IS-12437 Zirconium PowderDocument8 pagesIS-12437 Zirconium PowderAnuradhaPatraNo ratings yet
- Friday 15 January 2021: BiologyDocument20 pagesFriday 15 January 2021: BiologyAhmad ZaareerNo ratings yet
- 2 Ethyl-Hexanol Output Temperature Control in Hydrogenation ProcessDocument4 pages2 Ethyl-Hexanol Output Temperature Control in Hydrogenation ProcessShay BlueNo ratings yet
- Laboratory Drying Oven - Forced VentilationDocument8 pagesLaboratory Drying Oven - Forced VentilationALVES BROLIN MUCHA MALLAUPOMANo ratings yet
- RESPY Catalog (160411)Document2 pagesRESPY Catalog (160411)Haliun AltangerelNo ratings yet