You might also like
- AnemiaDocument41 pagesAnemiaBang FadNo ratings yet
- Electrolyte Imbalance, A Simple Guide To The Condition, Diagnosis, Treatment And Related ConditionsFrom EverandElectrolyte Imbalance, A Simple Guide To The Condition, Diagnosis, Treatment And Related ConditionsRating: 1 out of 5 stars1/5 (1)
- A Simple Guide to Hyperparathyroidism, Treatment and Related DiseasesFrom EverandA Simple Guide to Hyperparathyroidism, Treatment and Related DiseasesNo ratings yet
- Hematologic DisordersDocument30 pagesHematologic DisordersUday Kumar100% (1)
- Microcytic Hypochromic AnemiasDocument19 pagesMicrocytic Hypochromic Anemiassaket100% (2)
- Hyperkalemia, (High Blood Potassium) A Simple Guide To The Condition, Diagnosis, Treatment And Related ConditionsFrom EverandHyperkalemia, (High Blood Potassium) A Simple Guide To The Condition, Diagnosis, Treatment And Related ConditionsNo ratings yet
- Lecture 2-Blood RBC WBCDocument51 pagesLecture 2-Blood RBC WBCsamayaNo ratings yet
- Dr. THOMAS KINGSLEY MD Professor of Medicine TVMCDocument48 pagesDr. THOMAS KINGSLEY MD Professor of Medicine TVMCDr THOMAS KINGSLEY MDNo ratings yet
- Congestive Heart Failure PathophysiologyDocument7 pagesCongestive Heart Failure PathophysiologyAileen Grace RodrigoNo ratings yet
- 2021 ESC Preporuke Za Srčani Pejsing I Srčanu Resinhronizacionu TerapijuDocument68 pages2021 ESC Preporuke Za Srčani Pejsing I Srčanu Resinhronizacionu TerapijuDaria Lily MićanovićNo ratings yet
- Anemia in ChildrenDocument67 pagesAnemia in ChildrenAgung Maulana ArmstrongNo ratings yet
- AnemiaDocument63 pagesAnemiaRasoulNo ratings yet
- Bio Inorganic ChemistryDocument49 pagesBio Inorganic ChemistryMONIRUZZAMAN MONIRNo ratings yet
- Anemia Anak 2: Dr. BerthaDocument35 pagesAnemia Anak 2: Dr. BerthaAriNo ratings yet
- Anaemias: P. Manyau School of Pharmacy University of ZimbabweDocument41 pagesAnaemias: P. Manyau School of Pharmacy University of Zimbabwebrian mgabiNo ratings yet
- Functions and Properties of Blood - Plasma - Blood Cell Production - Erythrocytes - Blood Types - Leukocytes - Hemostasis (Stoppage of Bleeding)Document47 pagesFunctions and Properties of Blood - Plasma - Blood Cell Production - Erythrocytes - Blood Types - Leukocytes - Hemostasis (Stoppage of Bleeding)vanderphysNo ratings yet
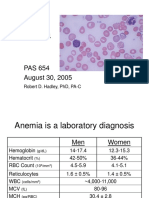
- Anemia Is A Laboratory Diagnosis: Men WomenDocument21 pagesAnemia Is A Laboratory Diagnosis: Men WomenOmaraye JoshuaNo ratings yet
- Chapter 2Document43 pagesChapter 2Puvaneswary SegharanNo ratings yet
- Red Cell DisordersDocument60 pagesRed Cell DisordersASMANI MATETENo ratings yet
- BLG111Document7 pagesBLG111Manasseh LawrenceNo ratings yet
- Microcytic Hypochromic Anemia: - M QariDocument33 pagesMicrocytic Hypochromic Anemia: - M QarirohitNo ratings yet
- LEC3Document26 pagesLEC3Jessica StewartNo ratings yet
- Anemia in ChildrenDocument33 pagesAnemia in ChildrenYemata HailuNo ratings yet
- Anemia Umam FazlurrahmanDocument44 pagesAnemia Umam Fazlurrahmanumam fazlurrahmanNo ratings yet
- Physiology of Blood: Ratna Kusumawati Department of Physiology, Faculty of Medicine Universitas Sebelas Maret SurakartaDocument65 pagesPhysiology of Blood: Ratna Kusumawati Department of Physiology, Faculty of Medicine Universitas Sebelas Maret SurakartaRizal AbdurrahmanNo ratings yet
- K9 - Iron Deficiency AnemiaDocument33 pagesK9 - Iron Deficiency AnemiaSiti FathiyaNo ratings yet
- Curs Hematologie An Feripriva EN Martie 2023 POSTATDocument55 pagesCurs Hematologie An Feripriva EN Martie 2023 POSTATErland BordNo ratings yet
- Hematinics (Iron and Vitamin B12)Document53 pagesHematinics (Iron and Vitamin B12)shareksultan5No ratings yet
- Case 11Document63 pagesCase 11حكيم العجلانNo ratings yet
- AnaemiasDocument60 pagesAnaemiasbvkjtzrvnyNo ratings yet
- Mod9Week2 - PA 3306 - Aug 28& Sept1Document46 pagesMod9Week2 - PA 3306 - Aug 28& Sept1komal sheikhNo ratings yet
- Blooddisorders 21 230610142333 f4472c75Document114 pagesBlooddisorders 21 230610142333 f4472c75Nabeel TahirNo ratings yet
- RBC DISORDERS Part I VR 2022Document63 pagesRBC DISORDERS Part I VR 2022victoria tavasNo ratings yet
- Hemtopoitic DrugDocument52 pagesHemtopoitic DrugUkash AbrahimNo ratings yet
- Clinical Hematological: Assist Prof. Dr. Mudhir S. ShekhaDocument31 pagesClinical Hematological: Assist Prof. Dr. Mudhir S. ShekhaAhmed. Masud.OthmanNo ratings yet
- 8 - Basics in Deficiency AnaemiasDocument59 pages8 - Basics in Deficiency AnaemiasWasana MendisNo ratings yet
- Hematopoiesis Anemia: Composition of The BloodDocument16 pagesHematopoiesis Anemia: Composition of The BloodOcha24 TupamahuNo ratings yet
- Biology Part-1 NotesDocument59 pagesBiology Part-1 NotesgjbxNo ratings yet
- Hypochromic AnemiasDocument51 pagesHypochromic AnemiasKaylee NesbitNo ratings yet
- Blood PharmacologyDocument71 pagesBlood PharmacologyNo NameNo ratings yet
- Haemopoiesis: Composition of Whole Blood & Its ComponentsDocument8 pagesHaemopoiesis: Composition of Whole Blood & Its ComponentsSafiya JamesNo ratings yet
- Topic 8 - Anemia 3Document62 pagesTopic 8 - Anemia 3Vince Martin ManaigNo ratings yet
- Anemia in ChildrenDocument67 pagesAnemia in ChildrenDenny BimatamaNo ratings yet
- Blood PhysiologyDocument17 pagesBlood Physiologymuhammad sadiqNo ratings yet
- Physiology of Blood: Ratna Kusumawati Department of Physiology, Faculty of Medicine Universitas Sebelas Maret SurakartaDocument60 pagesPhysiology of Blood: Ratna Kusumawati Department of Physiology, Faculty of Medicine Universitas Sebelas Maret SurakartaZidaneNo ratings yet
- Disease of The LiverDocument24 pagesDisease of The LiverMervis masatunyaNo ratings yet
- Anatomy and Physiology of BLOOD: Preet KaurDocument37 pagesAnatomy and Physiology of BLOOD: Preet KaurPreet KaurNo ratings yet
- Thalassaemia: Dr. Ruwanka de LiveraDocument30 pagesThalassaemia: Dr. Ruwanka de LiveraAshan BopitiyaNo ratings yet
- Obat AntianemiaDocument49 pagesObat AntianemiadilaNo ratings yet
- BloodDocument76 pagesBloodKerby Dela Fuente Alison100% (1)
- BloodDocument20 pagesBloodAdam PrabowoNo ratings yet
- Anemia in Childhood, Physiopathology and Clinical Findings Bleeding Tests in ChildhoodDocument76 pagesAnemia in Childhood, Physiopathology and Clinical Findings Bleeding Tests in Childhoodmohammed barwaryNo ratings yet
- Anaemia: Maekel Belay, MDDocument62 pagesAnaemia: Maekel Belay, MDjaffar sahilNo ratings yet
- Iron DeficiencyDocument42 pagesIron DeficiencyjanNo ratings yet
- Karpagcaseiron11Document34 pagesKarpagcaseiron11doctormpd28No ratings yet
- Disorders of Potassium Balance: Zhao Chenghai PathophysiologyDocument24 pagesDisorders of Potassium Balance: Zhao Chenghai PathophysiologyBayu SetiaNo ratings yet
- Interpreting Biochemistry Tutorial - StudentsDocument71 pagesInterpreting Biochemistry Tutorial - StudentsalrasatNo ratings yet
- 6.0 ElectrolytesDocument4 pages6.0 ElectrolytesHry WkNo ratings yet
- AnaemiaDocument108 pagesAnaemiaakoeljames8543No ratings yet
- BLOOD LecDocument46 pagesBLOOD LecGrape JuiceNo ratings yet
- Pharmacology NotesDocument22 pagesPharmacology NotesAlexia OwenNo ratings yet
- Fast Facts: Long-Chain Fatty Acid Oxidation Disorders for PatientsFrom EverandFast Facts: Long-Chain Fatty Acid Oxidation Disorders for PatientsNo ratings yet
- Physiology of Hemostasis - Dr. Rahmat Dani Satria PH.D., SP - PK (K)Document27 pagesPhysiology of Hemostasis - Dr. Rahmat Dani Satria PH.D., SP - PK (K)Viand NugrohoNo ratings yet
- AIIMS - Nov - 2012 - Paper @mmedicalbooksDocument289 pagesAIIMS - Nov - 2012 - Paper @mmedicalbooksbetsyNo ratings yet
- BIOLOGY FORM 4 Chapter 2 PDFDocument18 pagesBIOLOGY FORM 4 Chapter 2 PDFadele30No ratings yet
- Gram Staining Protocol or ProcedureDocument4 pagesGram Staining Protocol or Procedurearavindbt2523No ratings yet
- Dwnload Full Anatomy and Physiology 1st Edition Mckinley Solutions Manual PDFDocument35 pagesDwnload Full Anatomy and Physiology 1st Edition Mckinley Solutions Manual PDFleo7mco100% (17)
- Astrocyte: Structure & FunctionDocument7 pagesAstrocyte: Structure & Functionمحمود الموسويNo ratings yet
- Disorders of Red Blood CellsDocument27 pagesDisorders of Red Blood Cellskirti sharmaNo ratings yet
- Lecture Slides-01 02 Functional Microanatomy of NeuronsDocument4 pagesLecture Slides-01 02 Functional Microanatomy of NeuronsAna LeucaNo ratings yet
- Chist HidaticDocument25 pagesChist HidaticAna ScutelnicuNo ratings yet
- End Feel: K.SoundararajanDocument33 pagesEnd Feel: K.Soundararajanjack sparrowNo ratings yet
- Floyd17 TB Ch12Document5 pagesFloyd17 TB Ch12sdjuknicNo ratings yet
- Embryology Testis 08 PDFDocument20 pagesEmbryology Testis 08 PDFNanang Abdul AzizNo ratings yet
- Reproductive SystemDocument49 pagesReproductive SystemAyro Business CenterNo ratings yet
- RadiologyDocument1 pageRadiologyMark Christian BentijabaNo ratings yet
- 9B Respiration - Gaseous Exchange in Man PDFDocument18 pages9B Respiration - Gaseous Exchange in Man PDFabdulrehman mughalNo ratings yet
- Maternal ExamDocument20 pagesMaternal ExamChiz EscubuaNo ratings yet
- Cytophysiology Revised 2014Document19 pagesCytophysiology Revised 2014Julienne Sanchez-SalazarNo ratings yet
- Kuliah Biokimia-Imunokimia FK UNDIPDocument19 pagesKuliah Biokimia-Imunokimia FK UNDIPPutri HapsariNo ratings yet
- Health Psychology 9th Edition Taylor Test BankDocument25 pagesHealth Psychology 9th Edition Taylor Test BankTylerDavisbzmjy100% (64)
- ICO 2016 Basic Science Questions (1-40)Document9 pagesICO 2016 Basic Science Questions (1-40)Benjamin NgNo ratings yet
- Cervical and Shoulder 2020Document14 pagesCervical and Shoulder 2020Eduardo OlivaNo ratings yet
- Rain Natomy: Adapted From Human Anatomy & Physiology by Marieb and Hoehn (9 Ed.)Document16 pagesRain Natomy: Adapted From Human Anatomy & Physiology by Marieb and Hoehn (9 Ed.)vibs1994No ratings yet
- Torehj 6 1 PDFDocument20 pagesTorehj 6 1 PDFSaifuddin HaswareNo ratings yet
- Cephalometric Lecture-1Document45 pagesCephalometric Lecture-1Batman 02053No ratings yet
- Pteridophytes MorphologyDocument79 pagesPteridophytes MorphologyBoopathiAyyanarMNo ratings yet
- Life Processes in Living Organisms Part - 2 - 1Document21 pagesLife Processes in Living Organisms Part - 2 - 1Shyamal AryalNo ratings yet
- Experiment and Activity CellDocument4 pagesExperiment and Activity CellPang ChixxNo ratings yet
- Effects of Myofascial Release and Stretching Technique On ROM and Reaction Time PDFDocument3 pagesEffects of Myofascial Release and Stretching Technique On ROM and Reaction Time PDFfisionovaisNo ratings yet