You might also like
- Chapter 19 NMRDocument126 pagesChapter 19 NMRMuchammad RofiiNo ratings yet
- PH C NG Hư NG T H T Nhân Nuclear Magnetic ResonanceDocument68 pagesPH C NG Hư NG T H T Nhân Nuclear Magnetic ResonanceNguyễn ĐạtNo ratings yet
- Nuclear Magnetic Resonance Spectroscopy: Chem 8361/4361: Interpretation of Organic SpectraDocument70 pagesNuclear Magnetic Resonance Spectroscopy: Chem 8361/4361: Interpretation of Organic SpectraErizan AldiNo ratings yet
- 18MPH31C U4Document78 pages18MPH31C U4IISER MOHALINo ratings yet
- Chapter 8 - Nuclear Magnetic Resonance SpectrosDocument38 pagesChapter 8 - Nuclear Magnetic Resonance SpectrosBizuayehu Gero100% (1)
- NMR 1 PDFDocument120 pagesNMR 1 PDFbinseung skzNo ratings yet
- NMRDocument35 pagesNMRnikhila11reddyNo ratings yet
- NMRDocument17 pagesNMRSourabhNo ratings yet
- NMR Spectroscopy: Afsath. B Mpharm1 Year Pharmacognosy and Phytochemistry Malik Deenar College of PharmacyDocument23 pagesNMR Spectroscopy: Afsath. B Mpharm1 Year Pharmacognosy and Phytochemistry Malik Deenar College of PharmacychinmayeeNo ratings yet
- NMR Spectroscopy-ECEDocument77 pagesNMR Spectroscopy-ECEBhavya TanneruNo ratings yet
- Nuclear Magnetic Resonance (NMR) SpectrosDocument41 pagesNuclear Magnetic Resonance (NMR) SpectrosSivam AnandNo ratings yet
- NMR Spectroscopy For Structure ElucidationDocument47 pagesNMR Spectroscopy For Structure ElucidationSaw MyintNo ratings yet
- Nuclear Magnetic Resonance and Radiation TechniquesDocument15 pagesNuclear Magnetic Resonance and Radiation TechniquesMathavaraja JeyaramanNo ratings yet
- NMR SpectrosDocument42 pagesNMR SpectrosB1605 SAKSHAM MISHRANo ratings yet
- NMR-hands OnsDocument61 pagesNMR-hands OnsdillmannNo ratings yet
- NMR FinalDocument50 pagesNMR Finalpharmacologist786No ratings yet
- NMRDocument48 pagesNMRSREEHARI V NAIRNo ratings yet
- Nuclear Magnetic Resonance Spectroscopy: Absorption inDocument21 pagesNuclear Magnetic Resonance Spectroscopy: Absorption inFiona OyatsiNo ratings yet
- Nuclear Magnetic Resonance 1: Lecture Date: February 11, 2008Document40 pagesNuclear Magnetic Resonance 1: Lecture Date: February 11, 2008team engineerNo ratings yet
- NMR SpectrosDocument45 pagesNMR SpectrosfatemaNo ratings yet
- Topic 7 Nuclear Magnetic ResonanceDocument37 pagesTopic 7 Nuclear Magnetic ResonanceNurshuhada NordinNo ratings yet
- NMRDocument30 pagesNMRBharatula Suryamani Shankar ee19b013No ratings yet
- Organic ChemistryDocument101 pagesOrganic ChemistryHasithaNo ratings yet
- Fundamentals of Magnetic ResonancesadasdasdDocument42 pagesFundamentals of Magnetic ResonancesadasdasdtonikNo ratings yet
- NMR For BSCDocument13 pagesNMR For BSCmalluchaithra88_3587No ratings yet
- 5.Nmr Class LatestDocument81 pages5.Nmr Class Latestdimitra shenoyNo ratings yet
- Kuliah NMRDocument92 pagesKuliah NMRDedi saputraNo ratings yet
- NMR in Word FormDocument37 pagesNMR in Word FormsrideviNo ratings yet
- NMR (VP)Document59 pagesNMR (VP)Vishnu PriyaNo ratings yet
- Lecture 25 - RG - NMR - Chemical Shift - 7.10.2014Document30 pagesLecture 25 - RG - NMR - Chemical Shift - 7.10.2014Sampada DesaiNo ratings yet
- 1HNMR Lecture NotesDocument53 pages1HNMR Lecture NotesJian Hong Tee100% (1)
- LASERS: The Semiconductor Laser: Angel Valle (IFCA)Document33 pagesLASERS: The Semiconductor Laser: Angel Valle (IFCA)estaesladefinitivadedescargasNo ratings yet
- NMR - A Non Destructive Food Evaluation Technique: Ramesh. VDocument44 pagesNMR - A Non Destructive Food Evaluation Technique: Ramesh. VAnkit GoyalNo ratings yet
- B.Tech NMR RC Part 1 & 2Document68 pagesB.Tech NMR RC Part 1 & 2SricharanNo ratings yet
- Atomic SpectrosDocument129 pagesAtomic SpectrosmianNo ratings yet
- Electron Configurations and The Periodic Table: John W. Moore Conrad L. StanitskiDocument66 pagesElectron Configurations and The Periodic Table: John W. Moore Conrad L. StanitskiIvy RetuyaNo ratings yet
- 1D NMR SpectrosDocument28 pages1D NMR SpectrosAnggun AprilianiNo ratings yet
- NMR ReichiDocument186 pagesNMR ReichiJanni ManojkumarNo ratings yet
- Electron Spin Resonance SpectrosDocument56 pagesElectron Spin Resonance Spectrosboyapati venupriyaNo ratings yet
- NMR SpectrosDocument66 pagesNMR SpectrosDushyant Patel100% (1)
- Motion of The Atoms: Lattice VibrationsDocument11 pagesMotion of The Atoms: Lattice VibrationsBadri DadiNo ratings yet
- Magnetic Resonance: Compiled and Written By: Faith Van Nice 1992, Mark Haig Khachaturian 2002Document84 pagesMagnetic Resonance: Compiled and Written By: Faith Van Nice 1992, Mark Haig Khachaturian 2002Beautiful-Pretty Happy-WealthyNo ratings yet
- Atomic Structure: Kotz CH 7 & CH 22 (Sect 4,5)Document37 pagesAtomic Structure: Kotz CH 7 & CH 22 (Sect 4,5)Akhmad SafrinNo ratings yet
- CHME 222 - Lecture 6Document29 pagesCHME 222 - Lecture 6islam.lukmanov2003No ratings yet
- Raman ScatteringDocument65 pagesRaman ScatteringPragna ReddyNo ratings yet
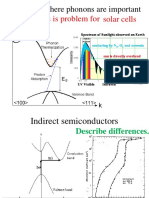
- Solar Cells Energy Loss Is Problem For: Examples Where Phonons Are ImportantDocument51 pagesSolar Cells Energy Loss Is Problem For: Examples Where Phonons Are Importantjose mirandaNo ratings yet
- AstroH SS10 NonthermalDocument28 pagesAstroH SS10 NonthermalHua Hidari YangNo ratings yet
- NMR Info Tables 12-31-09Document48 pagesNMR Info Tables 12-31-09NahdaNo ratings yet
- Atomic Term SymbolDocument13 pagesAtomic Term SymbolUmendra KhokharNo ratings yet
- NMR PPDocument28 pagesNMR PPparvaneh.eNo ratings yet
- Notes 05 HMR v26 Part1Document38 pagesNotes 05 HMR v26 Part1cj AndersonNo ratings yet
- NMR Spectroscopy: The TheoryDocument4 pagesNMR Spectroscopy: The Theoryoliv1aNo ratings yet
- Fundamentals of Radio AstronomyDocument23 pagesFundamentals of Radio AstronomyPriya WaghelaNo ratings yet
- Nuclear Magnetic Resonance SpectrosDocument74 pagesNuclear Magnetic Resonance Spectrosrejie magnayeNo ratings yet
- Electronic Structure of Atoms - General ChemistryDocument92 pagesElectronic Structure of Atoms - General ChemistryDuc Anh NguyenNo ratings yet
- ch1 - Review of QEDocument21 pagesch1 - Review of QEfuyoungNo ratings yet
- Matthews - Biological NMR - L1Document13 pagesMatthews - Biological NMR - L1AlexTsuiNo ratings yet
- Electronic Devices and Circuits: The Commonwealth and International Library: Electrical Engineering Division, Volume 1From EverandElectronic Devices and Circuits: The Commonwealth and International Library: Electrical Engineering Division, Volume 1Rating: 4.5 out of 5 stars4.5/5 (5)
- Electronic Devices and Circuits: The Commonwealth and International Library: Electrical Engineering Division, Volume 3From EverandElectronic Devices and Circuits: The Commonwealth and International Library: Electrical Engineering Division, Volume 3Rating: 3 out of 5 stars3/5 (2)
- Advances in Structure Research by Diffraction Methods: Fortschritte der Strukturforschung mit BeugungsmethodenFrom EverandAdvances in Structure Research by Diffraction Methods: Fortschritte der Strukturforschung mit BeugungsmethodenR. BrillNo ratings yet
- Dandelion Seeds Blowing PowerPoint TemplatesDocument48 pagesDandelion Seeds Blowing PowerPoint TemplatesSanjay GuptaNo ratings yet
- 1) Cpm/Pert Network For Moore House Contractors (See Paper)Document3 pages1) Cpm/Pert Network For Moore House Contractors (See Paper)Eudes Salvy AssoumouNo ratings yet
- Daftar Siswa Kelas Xi DG 2Document33 pagesDaftar Siswa Kelas Xi DG 2yusuframdhani91No ratings yet
- Ima 22-23Document4 pagesIma 22-23Archak SinghNo ratings yet
- EVM-Calculation TemplateDocument5 pagesEVM-Calculation TemplatePavethraNo ratings yet
- CHE S402 Chapter 4 Solid Catalysts Part2Document9 pagesCHE S402 Chapter 4 Solid Catalysts Part2Rashmi SahooNo ratings yet
- MRP Dan MRP Ii: Departemen ProduksiDocument48 pagesMRP Dan MRP Ii: Departemen ProduksiYulia KusumantiNo ratings yet
- Lab Report 2Document12 pagesLab Report 2Fatihah AlhataNo ratings yet
- Plantilla Powerpoint de Metodos AnticonceptivosDocument50 pagesPlantilla Powerpoint de Metodos AnticonceptivosPatty MoralesNo ratings yet
- Project ManagementDocument2 pagesProject ManagementAlishba NadeemNo ratings yet
- Edexcel Chemistry A-Level: Topic 19: Modern Analytical Techniques IIDocument8 pagesEdexcel Chemistry A-Level: Topic 19: Modern Analytical Techniques IILulwa KhaskiehNo ratings yet
- Global Logistics Network PowerPointDocument45 pagesGlobal Logistics Network PowerPointEnjiniring UPK TamboraNo ratings yet
- Bab Ix Sistem Informasi Jasa Kon Struksi: Virgo Ray Labams NaibahoDocument34 pagesBab Ix Sistem Informasi Jasa Kon Struksi: Virgo Ray Labams NaibahoVirgo Ray NaibahoNo ratings yet
- Pencil Ladder Shadow PowerPoint TemplatesDocument48 pagesPencil Ladder Shadow PowerPoint TemplatesFebrian SaputraNo ratings yet
- Plantilla PowerPoint de Primeros AuxiliosDocument45 pagesPlantilla PowerPoint de Primeros AuxiliosPatty MoralesNo ratings yet
- Padang Rumput GeoDocument48 pagesPadang Rumput GeoKenieNo ratings yet
- Analysing A Photographer - Man RayDocument1 pageAnalysing A Photographer - Man Rayapi-563918643No ratings yet
- 14 - Back To School Education TemplateDocument38 pages14 - Back To School Education TemplatePanca NababanNo ratings yet
- Chapter 1 - Perspective DrawingDocument23 pagesChapter 1 - Perspective DrawingChe Nora Che HassanNo ratings yet
- Characterization of NanoparticlesDocument16 pagesCharacterization of NanoparticlesPrasun SarkarNo ratings yet
- GCSE YR 10 Photography WEEK 3 (B) 20th - 24th SepDocument10 pagesGCSE YR 10 Photography WEEK 3 (B) 20th - 24th SepElla DGNo ratings yet
- Lec 7 - Intro To CryptographyDocument38 pagesLec 7 - Intro To CryptographygNo ratings yet
- Free Online Library PPT Templates: Insert The Subtitle of Your PresentationDocument48 pagesFree Online Library PPT Templates: Insert The Subtitle of Your PresentationArizona Hantaran Mahar JambiNo ratings yet
- PCR-laboratorij Za Molekularna Genetička Ispitivanja: Datum rođenja/Birthdate/GeburtsdatumDocument1 pagePCR-laboratorij Za Molekularna Genetička Ispitivanja: Datum rođenja/Birthdate/GeburtsdatumDžana DahićNo ratings yet
- What Is HPLC Column Conditioning - How Long To EquilibrateDocument5 pagesWhat Is HPLC Column Conditioning - How Long To EquilibrateMubarak PatelNo ratings yet
- Analytical Method of Chlorfenapyr in SCDocument2 pagesAnalytical Method of Chlorfenapyr in SCMeilaDwiputriNo ratings yet
- Catalyst Characterization 2Document37 pagesCatalyst Characterization 2Mo MobarkNo ratings yet
- Emwas Et Al. - 2019 - NMR Spectroscopy For Metabolomics ResearchDocument39 pagesEmwas Et Al. - 2019 - NMR Spectroscopy For Metabolomics Researchyannick brunatoNo ratings yet
- FORINSTDocument7 pagesFORINSTYoMomma PinkyNo ratings yet
- Cara Menggunakan MikroskopDocument7 pagesCara Menggunakan Mikroskopmetry todingNo ratings yet