You might also like
- Cardiovascular PathologyDocument133 pagesCardiovascular Pathologymulugetaketema394No ratings yet
- Atherosclerosis: CH ApterDocument22 pagesAtherosclerosis: CH ApterAnonymous mmA06fqNEdNo ratings yet
- Atherosklerosis, Artheriosklerosis Dan Kelainan VaskulerDocument66 pagesAtherosklerosis, Artheriosklerosis Dan Kelainan VaskulerOlivia Chandra DeviNo ratings yet
- Vascular Endothelium e Gatekeeper of Vessel Health: AtherosclerosisDocument17 pagesVascular Endothelium e Gatekeeper of Vessel Health: AtherosclerosisLia FitriaNo ratings yet
- Lecture13 & 14aDocument5 pagesLecture13 & 14aIsak ShatikaNo ratings yet
- Arterial and Venous PathologyDocument93 pagesArterial and Venous PathologyAmanuel MaruNo ratings yet
- Arterial and Venous PathologyDocument93 pagesArterial and Venous PathologyAmanuel Maru100% (2)
- Cvs Lec 1Document43 pagesCvs Lec 1data4fourgNo ratings yet
- Chapter 11 Blood Vessels 8th Ed NotesDocument7 pagesChapter 11 Blood Vessels 8th Ed NotesKyle Christopher SiaNo ratings yet
- Atherosclerotic Process and The Risk Factors: Dr. Budiana Tanurahardja.,Sp - Pa Dr. Lisnawati SppaDocument34 pagesAtherosclerotic Process and The Risk Factors: Dr. Budiana Tanurahardja.,Sp - Pa Dr. Lisnawati SppaPatrisius BonggalinoqNo ratings yet
- Essential of Special Patholog by DR Zair Hassan: December 2015Document271 pagesEssential of Special Patholog by DR Zair Hassan: December 2015pdf pediatriNo ratings yet
- The Pathophysiology of Acute Coronary SyndromesDocument6 pagesThe Pathophysiology of Acute Coronary SyndromesTulisan NonaNo ratings yet
- 2 - AtherosclerosisDocument16 pages2 - AtherosclerosisUnnati PatelNo ratings yet
- Semana 6 AteroesclerosisDocument5 pagesSemana 6 AteroesclerosisrobertoNo ratings yet
- SG 4,5,6Document20 pagesSG 4,5,6Itadori YujiNo ratings yet
- Patogenesis AterosklerosisDocument3 pagesPatogenesis AterosklerosisMarsella Epifania SuwignyoNo ratings yet
- Lili AteroDocument2 pagesLili AteroDavy JonesNo ratings yet
- Vascular Biology of Atherosclerosis Normal Arterial WallDocument2 pagesVascular Biology of Atherosclerosis Normal Arterial WallDavy JonesNo ratings yet
- DB13 - Pathophysiology of AtherosclerosisDocument2 pagesDB13 - Pathophysiology of Atherosclerosisi_vhie03No ratings yet
- FCVM 08 750510Document11 pagesFCVM 08 750510Joseph SebastianNo ratings yet
- Pathology of Blood VesselsDocument80 pagesPathology of Blood VesselsiqiqiqiqiqNo ratings yet
- COVID 19 Associated Vasculitis and Vasculopathy: Richard C. BeckerDocument13 pagesCOVID 19 Associated Vasculitis and Vasculopathy: Richard C. Beckerbaba ababNo ratings yet
- Building Bodies 2Document20 pagesBuilding Bodies 2xqm45552j6No ratings yet
- Tinywow - L.3 Condorelli Atherosclerosis - 28139364Document14 pagesTinywow - L.3 Condorelli Atherosclerosis - 28139364filymascoloNo ratings yet
- Necrosis NotesDocument3 pagesNecrosis Notesjerad19No ratings yet
- ATHEROMA What is an AtheromaDocument1 pageATHEROMA What is an AtheromaetengNo ratings yet
- Repair After Myocardial Infarction, Between Fantasy and RealityDocument6 pagesRepair After Myocardial Infarction, Between Fantasy and RealitydanielcpnkimoNo ratings yet
- To Remember The Four Causes of Cell InjuryDocument43 pagesTo Remember The Four Causes of Cell Injuryapi-3825096No ratings yet
- Pathophysiology of Atherosclerosis 1Document26 pagesPathophysiology of Atherosclerosis 1jackNo ratings yet
- Pathology EssayDocument19 pagesPathology EssayARUNSKNo ratings yet
- Atherosclerosis: Inflammation & Oxidative StressDocument13 pagesAtherosclerosis: Inflammation & Oxidative StressDarlene Shaira JuntillaNo ratings yet
- The Pathophysiology of Acute Coronary SyndromesDocument7 pagesThe Pathophysiology of Acute Coronary SyndromesPrita RosdianaNo ratings yet
- Atherosclerosis: Jana NovotnáDocument28 pagesAtherosclerosis: Jana Novotnásunita_aug801No ratings yet
- PATHOGENESIS, RISK FACTORS AND PREVENTION OF ATHEROSCLEROSISDocument5 pagesPATHOGENESIS, RISK FACTORS AND PREVENTION OF ATHEROSCLEROSISastalithalorelNo ratings yet
- Cells 10 00226Document26 pagesCells 10 00226spraptamaNo ratings yet
- 2.risk Factors & Pathogenesis of AtherosclerosisDocument18 pages2.risk Factors & Pathogenesis of Atherosclerosismasa.dalati.1No ratings yet
- Rutherford: Vascular Surgery, 6th EdDocument6 pagesRutherford: Vascular Surgery, 6th EdanitaromeroNo ratings yet
- Cardiovascular SystemDocument22 pagesCardiovascular SystemWadabiNo ratings yet
- Pathogenesis of Atherosclerosis: These Are The Composition of YourDocument4 pagesPathogenesis of Atherosclerosis: These Are The Composition of Yourchocoholic potchiNo ratings yet
- Pathology Week9 BloodVesselsDocument4 pagesPathology Week9 BloodVesselsSalifyanji SimpambaNo ratings yet
- Atherosclerosis SummaryDocument3 pagesAtherosclerosis SummaryaliceNo ratings yet
- Nutrition and AtherosclerosisDocument19 pagesNutrition and AtherosclerosisJOSE RICARDO LOPEZ PALESTINANo ratings yet
- Coronary Artery AtherosclerosisDocument25 pagesCoronary Artery AtherosclerosisShahrizal Che JamelNo ratings yet
- Cardiovascular Pathology 2024Document132 pagesCardiovascular Pathology 2024Chen HouyuNo ratings yet
- Atherosclerosis. Biochemical Modifications in Acute Coronary SyndromeDocument12 pagesAtherosclerosis. Biochemical Modifications in Acute Coronary Syndromej.doe.hex_87No ratings yet
- AtherosclerosisDocument36 pagesAtherosclerosisjainilNo ratings yet
- Cardiovascular DentDocument92 pagesCardiovascular DentVincent De AsisNo ratings yet
- Cardiovascular PathologyDocument35 pagesCardiovascular Pathologyzeroun2480% (5)
- Patologi Bedah Sistem Vaskuler A Dan VDocument77 pagesPatologi Bedah Sistem Vaskuler A Dan Vfienda feraniNo ratings yet
- NECROSIS - causes, types and morphological signsDocument5 pagesNECROSIS - causes, types and morphological signsBeatrice BeezusNo ratings yet
- Lecture 7 - Atherosclerosis and Ischemic Heart DiseaseDocument39 pagesLecture 7 - Atherosclerosis and Ischemic Heart Diseasemayag3243No ratings yet
- Blood Vessel Structure and Function GuideDocument8 pagesBlood Vessel Structure and Function GuideshinbiNo ratings yet
- Neuro PathologyDocument363 pagesNeuro PathologyGvanca IordanishviliNo ratings yet
- 10.1038@s41581 018 0098 ZDocument22 pages10.1038@s41581 018 0098 Zwora123potNo ratings yet
- 3-Atherosclerosis and Acute Limb IschemiaDocument17 pages3-Atherosclerosis and Acute Limb Ischemiaوهيب الجعفريNo ratings yet
- Inflammation overviewDocument10 pagesInflammation overviewYaff DthNo ratings yet
- Vascular Pathology - Lesson - 10Document81 pagesVascular Pathology - Lesson - 10Aya AhmedNo ratings yet
- The Cardiovascular Atherosclerotic Disease Is One of The Causes of Mortality and Economic BurdenDocument14 pagesThe Cardiovascular Atherosclerotic Disease Is One of The Causes of Mortality and Economic BurdenBryantNo ratings yet
- Microcirculation in Cardiovascular DiseasesFrom EverandMicrocirculation in Cardiovascular DiseasesEnrico Agabiti-RoseiNo ratings yet
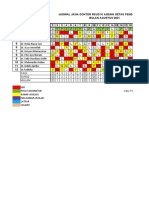
- Jadwal Jaga Dokter RSUD Ki Ageng Getas Pendowo GubugDocument3 pagesJadwal Jaga Dokter RSUD Ki Ageng Getas Pendowo GubugArdian AshadiNo ratings yet
- Phagocytosis A Fundamental Process in ImmunityDocument19 pagesPhagocytosis A Fundamental Process in ImmunityMarco MPNo ratings yet
- HHS Public AccessDocument13 pagesHHS Public AccessArdian AshadiNo ratings yet
- Shah 2000Document4 pagesShah 2000Ardian AshadiNo ratings yet
- 2019ltr PDFDocument13 pages2019ltr PDFMarlon Quispe MatiasNo ratings yet
- Pi Is 2214109 X 17302930Document10 pagesPi Is 2214109 X 17302930Anonymous 2fAXhpNo ratings yet
- Nutrients 12 00878Document13 pagesNutrients 12 00878Ardian AshadiNo ratings yet
- Pi Is 2214109 X 17302930Document10 pagesPi Is 2214109 X 17302930Anonymous 2fAXhpNo ratings yet
- Pone 0167708Document10 pagesPone 0167708Ardian AshadiNo ratings yet
- Preserving Healthy Muscle During Weight Loss: Review From Asn Eb 2016 SymposiumDocument9 pagesPreserving Healthy Muscle During Weight Loss: Review From Asn Eb 2016 Symposiumramsol07No ratings yet
- Pone 0233118Document12 pagesPone 0233118Ardian AshadiNo ratings yet
- Nutrients 12 00878Document13 pagesNutrients 12 00878Ardian AshadiNo ratings yet
- Game TheoryDocument21 pagesGame TheoryJoan Albet SerraNo ratings yet
- Nutrients 10 00108Document11 pagesNutrients 10 001084444alisaNo ratings yet
- Fasting For Weight Loss 41Document7 pagesFasting For Weight Loss 41Mikaella Ninochka Custodio PelayoNo ratings yet
- A Guide To Advancing Your Career With Essential Business SkillsDocument16 pagesA Guide To Advancing Your Career With Essential Business SkillsArdian AshadiNo ratings yet
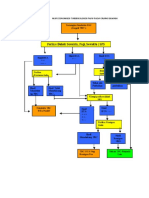
- Alur Diagnosis TBC 1Document1 pageAlur Diagnosis TBC 1Ardian AshadiNo ratings yet
- Presentation 1Document3 pagesPresentation 1Ardian AshadiNo ratings yet
- Journal Reading Neuro enDocument7 pagesJournal Reading Neuro enArdian AshadiNo ratings yet
- Neutralizing IL-17 Suppresses Allergic Rhinitis by Modulating Immune ResponsesDocument9 pagesNeutralizing IL-17 Suppresses Allergic Rhinitis by Modulating Immune ResponsesArdian AshadiNo ratings yet
- Management of KeratitisDocument12 pagesManagement of KeratitisSeptian Harry WibowoNo ratings yet
- Radiation Dose Optimization For The Bolus Tracking Technique in Abdominal Computed Tomography: Usefulness of Real-Time Iterative Reconstruction For Monitoring ScanDocument6 pagesRadiation Dose Optimization For The Bolus Tracking Technique in Abdominal Computed Tomography: Usefulness of Real-Time Iterative Reconstruction For Monitoring ScanArdian AshadiNo ratings yet
- Oxygen-Ozone Therapy in The Treatment of Adipose TissueDocument9 pagesOxygen-Ozone Therapy in The Treatment of Adipose Tissueweb3351100% (1)
- Diaz Flores 32 1239 1279 2017Document41 pagesDiaz Flores 32 1239 1279 2017María P SNo ratings yet
- Atherosclerosis PathophysiologyDocument11 pagesAtherosclerosis PathophysiologyCandice ChengNo ratings yet
- Biotechnology Cell Laboratory Manager in Boston MA Resume Neil TritmanDocument3 pagesBiotechnology Cell Laboratory Manager in Boston MA Resume Neil TritmanNeilTritmanNo ratings yet
- Pathogenesis of Malarial ComplicationsDocument9 pagesPathogenesis of Malarial ComplicationsErrold Joseph LahaganNo ratings yet
- Von Willebrand Factor and Endothelial Damage: A Possible Association With Covid-19Document4 pagesVon Willebrand Factor and Endothelial Damage: A Possible Association With Covid-19Rehabilitasi Narkoba Ar RahmanNo ratings yet
- Neurochemistry International: Satoshi Hosoki, Tomotaka Tanaka, Masafumi IharaDocument9 pagesNeurochemistry International: Satoshi Hosoki, Tomotaka Tanaka, Masafumi IharaindanazulfaaNo ratings yet
- Long COVID Major Findings Mechanisms Recommendations-2023Document14 pagesLong COVID Major Findings Mechanisms Recommendations-2023Zezinho zéNo ratings yet
- Veterinarian Resume for Trisha LiuDocument2 pagesVeterinarian Resume for Trisha LiuZhishanNo ratings yet
- Lymphangiogenesis in Cancer Metastasis by Dr. Steven A. Stacker, Dr. Marc G. Achen (Auth.)Document255 pagesLymphangiogenesis in Cancer Metastasis by Dr. Steven A. Stacker, Dr. Marc G. Achen (Auth.)wei wangNo ratings yet
- Full Download Ebook Ebook PDF Ocular Pathology 8th Edition by Yanoff MD Myron PDFDocument23 pagesFull Download Ebook Ebook PDF Ocular Pathology 8th Edition by Yanoff MD Myron PDFjoanne.hines504100% (38)
- Download ebook Vascular Medicine A Companion To Braunwalds Heart Disease Pdf full chapter pdfDocument67 pagesDownload ebook Vascular Medicine A Companion To Braunwalds Heart Disease Pdf full chapter pdfelsie.mcintyre883100% (24)
- Expert Recommendations On The Assessment of Wall Shear Stress in Human Coronary ArteriesDocument17 pagesExpert Recommendations On The Assessment of Wall Shear Stress in Human Coronary ArteriesМихаил НеболеевNo ratings yet
- Fibrinolytic System and Anticoagulants: Fathima.ADocument15 pagesFibrinolytic System and Anticoagulants: Fathima.AFATHIMA ANo ratings yet
- Trauma Induced Coagulopathy What You Need To Know.1Document7 pagesTrauma Induced Coagulopathy What You Need To Know.1llucaspilonettoNo ratings yet
- Circulatory system microanatomyDocument5 pagesCirculatory system microanatomyJohnny BravoNo ratings yet
- Robbins Basic Pathology 10th Edition-Pages-67-105Document45 pagesRobbins Basic Pathology 10th Edition-Pages-67-105newtihstishNo ratings yet
- Pulmonary Hypertension: A Guide to Pathogenesis, Diagnosis and TreatmentDocument27 pagesPulmonary Hypertension: A Guide to Pathogenesis, Diagnosis and Treatmentantonello picernoNo ratings yet
- 5 - HealingDocument29 pages5 - HealingShna SaadiNo ratings yet
- Artritis HipertrofikDocument25 pagesArtritis Hipertrofikadmin reumatologiNo ratings yet
- Sepsis, Pathophysiology and Clinical Management (BMJ, 2016)Document20 pagesSepsis, Pathophysiology and Clinical Management (BMJ, 2016)Juan Luis Nuñez AravenaNo ratings yet
- Principles and Pitfalls in Coronary Vasomotor Function TestingDocument26 pagesPrinciples and Pitfalls in Coronary Vasomotor Function TestingRamón López PalopNo ratings yet
- Neuroprotective Effects of Ginkgo Biloba ExtractDocument14 pagesNeuroprotective Effects of Ginkgo Biloba Extractcpt-n3moNo ratings yet
- Endothelial Dysfunction in Diabetic Erectile DysfunctionDocument11 pagesEndothelial Dysfunction in Diabetic Erectile DysfunctionILham SyahNo ratings yet
- Curier Med Pag 51 PDFDocument80 pagesCurier Med Pag 51 PDFJardan TatianaNo ratings yet
- Circulation of the Pulp MicrocirculationDocument3 pagesCirculation of the Pulp MicrocirculationKarina HernandezNo ratings yet
- Acid UricDocument3 pagesAcid UricCosmin AndreiNo ratings yet
- INFLAMMATION: A Protective Response to InjuryDocument22 pagesINFLAMMATION: A Protective Response to InjuryRamesh KumarNo ratings yet
- Human Biological Science 2 BIOL122: National Workshop Manual 2019Document78 pagesHuman Biological Science 2 BIOL122: National Workshop Manual 2019Huge Lovely SmileNo ratings yet
- The Neuronal EnvironmentDocument428 pagesThe Neuronal EnvironmentPandaNo ratings yet