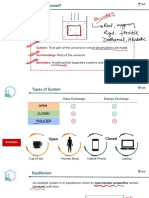
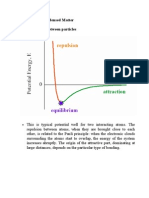
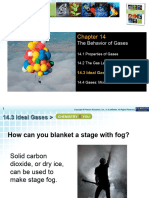
You might also like
- Properties of Gases (Report)Document19 pagesProperties of Gases (Report)Rex LapisNo ratings yet
- CHY 111 Unit 2 Thermo Lecture 1Document20 pagesCHY 111 Unit 2 Thermo Lecture 1Keshav ChandakNo ratings yet
- Physical Chemistry: Chemical EngineeringDocument11 pagesPhysical Chemistry: Chemical EngineeringEd Ryan RualesNo ratings yet
- Chemical Engineering Academic Reinforcement Sessions: CHE216: Physical Chemistry For Engineers 1Document7 pagesChemical Engineering Academic Reinforcement Sessions: CHE216: Physical Chemistry For Engineers 1Andrew OracionNo ratings yet
- 1 Intro Gases THermodynamics 2022Document15 pages1 Intro Gases THermodynamics 2022Jey BlaQNo ratings yet
- Three-Parameter Cubic Equation of State For Normal SubstancesDocument4 pagesThree-Parameter Cubic Equation of State For Normal SubstancesFiorela VillalobosNo ratings yet
- Thermal Physics Equations: 1. Ideal Gas LawDocument6 pagesThermal Physics Equations: 1. Ideal Gas LawThanh NgânNo ratings yet
- Physical ChemDocument57 pagesPhysical ChemDENISE COLENo ratings yet
- Introduction To Physical ChemistryDocument42 pagesIntroduction To Physical ChemistryRheanne SantosNo ratings yet
- 16 Komposisi Z RealGas Pseudocrtical Katz KayRuleDocument51 pages16 Komposisi Z RealGas Pseudocrtical Katz KayRulelarasatikarlindaNo ratings yet
- Physical Chemistry: Concepts and ApplicationsDocument25 pagesPhysical Chemistry: Concepts and Applicationszeeshanahmad111No ratings yet
- Introduction To Chemical Engineering Thermodynamics: Prepared by A. R. Caparanga, PHD For Ch126PDocument35 pagesIntroduction To Chemical Engineering Thermodynamics: Prepared by A. R. Caparanga, PHD For Ch126PDOZPandaNo ratings yet
- Chem 105-8, Sections 8.1, 8.2, 8.5 NotesDocument36 pagesChem 105-8, Sections 8.1, 8.2, 8.5 NotesKhang TrầnNo ratings yet
- Homework TermodinamicaDocument4 pagesHomework Termodinamicacarlosprez212No ratings yet
- Physical ChemistryDocument43 pagesPhysical Chemistrydrami94No ratings yet
- Basic Thermodynamics: Module OrientationDocument12 pagesBasic Thermodynamics: Module OrientationBenjie flor CalayegNo ratings yet
- 1 Intro Gases THermodynamicsDocument16 pages1 Intro Gases THermodynamicsThabiso GwijiNo ratings yet
- Single Phase Diagram - Real Gases - 003Document36 pagesSingle Phase Diagram - Real Gases - 003JatskinesisNo ratings yet
- CHEMICAL THERMODYNAMICS-III (1) 1st SlideDocument41 pagesCHEMICAL THERMODYNAMICS-III (1) 1st SlideMuhammadObaidullahNo ratings yet
- Sep08 PL2Document3 pagesSep08 PL2eka123No ratings yet
- CHY 111 Thermo Lecture 1Document19 pagesCHY 111 Thermo Lecture 1Nagarajan RathnakumarNo ratings yet
- l1 Thermo 23Document161 pagesl1 Thermo 23leomessi73870No ratings yet
- Gases and Other Properties: Lesson 5Document7 pagesGases and Other Properties: Lesson 5lucifer angelNo ratings yet
- Gas TheoryDocument65 pagesGas TheoryNicole OssaNo ratings yet
- NMAT Review 2018 Module - ChemistryDocument16 pagesNMAT Review 2018 Module - ChemistryRaf Lin DrawsNo ratings yet
- Thermodynamics: © Matthew W. MilliganDocument32 pagesThermodynamics: © Matthew W. MilliganAnton KortenkampNo ratings yet
- Lecture 12Document5 pagesLecture 12saadi yusufNo ratings yet
- Actual Molecular Mass Empirical Molecular Mass: Che 102 Chemistry For Engineers ReviewDocument8 pagesActual Molecular Mass Empirical Molecular Mass: Che 102 Chemistry For Engineers ReviewTariq Ceniza RasulNo ratings yet
- Vle 1Document40 pagesVle 1jeff.she1811No ratings yet
- L2 States of MatterDocument53 pagesL2 States of MatterAaryan ChodankarNo ratings yet
- ME 300 Div1 Lecture 01Document22 pagesME 300 Div1 Lecture 01Halil İbrahim KüplüNo ratings yet
- Unit 4 Chemistry NotesDocument72 pagesUnit 4 Chemistry NotesAsma AkterNo ratings yet
- Introduction To The Course: Termodinamika Dan Fisika Statistik - FIS62113Document121 pagesIntroduction To The Course: Termodinamika Dan Fisika Statistik - FIS62113rahmadani fitrianaNo ratings yet
- The Properties of Gases - CH 1Document14 pagesThe Properties of Gases - CH 1Tiara ElsinitaNo ratings yet
- Phases DiagramsDocument97 pagesPhases DiagramsSidharth JessyNo ratings yet
- Physical Chemistry Establishes and Develops The: Pchem I 1.1Document26 pagesPhysical Chemistry Establishes and Develops The: Pchem I 1.1Kaaya GodfreyNo ratings yet
- Applications of Differential EquationsDocument9 pagesApplications of Differential EquationsMuhammad AkhtarNo ratings yet
- AE 411 Prelim Module 1 - Review of Basic Thermodynamics PDFDocument15 pagesAE 411 Prelim Module 1 - Review of Basic Thermodynamics PDFKen OracioNo ratings yet
- MECH 325 Notes April 2004Document108 pagesMECH 325 Notes April 2004dNo ratings yet
- CHEM103 LW4 AnnotatedDocument25 pagesCHEM103 LW4 AnnotatedOmar MatarNo ratings yet
- Ilovepdf Merged RemovedDocument144 pagesIlovepdf Merged RemovedRajeshNo ratings yet
- CH 6Document19 pagesCH 6Ignas ŠakuroNo ratings yet
- 2022 - Chem 7023 - LectureNotes - 0824-1Document15 pages2022 - Chem 7023 - LectureNotes - 0824-1Uche PNo ratings yet
- THERMODYNAMICS 1 Rev 2Document62 pagesTHERMODYNAMICS 1 Rev 2Jads Cayabyab100% (2)
- Gas LawsDocument66 pagesGas LawsLorilieNo ratings yet
- Chapter 1 - Chemical Equilibrium Part 1Document36 pagesChapter 1 - Chemical Equilibrium Part 1Ng Kee NainNo ratings yet
- Chapter 1 2 - DR JalalDocument67 pagesChapter 1 2 - DR JalalScorpion RoyalNo ratings yet
- First Law of ThermodynamicsDocument107 pagesFirst Law of ThermodynamicsAmante Rivera Jr100% (1)
- Thermo - 1 (21-22) B&CDocument45 pagesThermo - 1 (21-22) B&CmsksksNo ratings yet
- Thermo T3Document15 pagesThermo T3Galilee SamuelsNo ratings yet
- Zero LawDocument10 pagesZero Lawalaa anwarNo ratings yet
- Chapter 03 Volumetric Properties of Pure Fluids 4 Slides Per PageDocument8 pagesChapter 03 Volumetric Properties of Pure Fluids 4 Slides Per PageHana Atalia100% (1)
- Dynamic MethodsDocument62 pagesDynamic Methodspainternetmx1No ratings yet
- 1 Chemical Engineering Thermodynamics IDocument37 pages1 Chemical Engineering Thermodynamics IYashib KamranNo ratings yet
- Problem 2725Document29 pagesProblem 2725Clyde SuerteNo ratings yet
- Chapter 1Document5 pagesChapter 1nisa_121babypinkNo ratings yet
- MCAT ChemistryDocument3 pagesMCAT ChemistryDurvPatelNo ratings yet
- Worked Problems in Heat, Thermodynamics and Kinetic Theory for Physics Students: The Commonwealth and International Library: Physics DivisionFrom EverandWorked Problems in Heat, Thermodynamics and Kinetic Theory for Physics Students: The Commonwealth and International Library: Physics DivisionRating: 4 out of 5 stars4/5 (3)
- Practice Makes Perfect in Chemistry: The Physical Behavior of MatterFrom EverandPractice Makes Perfect in Chemistry: The Physical Behavior of MatterRating: 5 out of 5 stars5/5 (1)
- Day 08Document7 pagesDay 08احمد الدلالNo ratings yet
- Lecture 6 1-29-2004Document27 pagesLecture 6 1-29-2004احمد الدلالNo ratings yet
- Topic 4 The Energy Balance 05Document10 pagesTopic 4 The Energy Balance 05احمد الدلالNo ratings yet
- Fluids 10Document22 pagesFluids 10احمد الدلالNo ratings yet
- Exam0102 DasrbitsDocument4 pagesExam0102 Dasrbitsاحمد الدلالNo ratings yet
- Chapter 10Document9 pagesChapter 10احمد الدلالNo ratings yet
- Elementary Principles of Chemical Processes 3 (1) - 655-681Document27 pagesElementary Principles of Chemical Processes 3 (1) - 655-681arnoldNo ratings yet
- Day 05Document10 pagesDay 05احمد الدلالNo ratings yet
- Integral Relations - Part 2Document23 pagesIntegral Relations - Part 2احمد الدلالNo ratings yet
- Lecture #2-Physical Chemistry - 8 (L2-P-2)Document10 pagesLecture #2-Physical Chemistry - 8 (L2-P-2)احمد الدلالNo ratings yet
- Fluids 3Document11 pagesFluids 3احمد الدلالNo ratings yet
- J Jfoodeng 2007 11 020Document12 pagesJ Jfoodeng 2007 11 020احمد الدلالNo ratings yet
- EBMF4103 (Chapter 1) Fluid Mechanics For Mechanical EngineeringDocument32 pagesEBMF4103 (Chapter 1) Fluid Mechanics For Mechanical EngineeringMahmud AbdullahNo ratings yet
- Chem12 C14 L3 LoDocument51 pagesChem12 C14 L3 Loاحمد الدلالNo ratings yet
- Section 13 HaywardDocument26 pagesSection 13 Haywardاحمد الدلالNo ratings yet
- Chapter 12Document27 pagesChapter 12احمد الدلالNo ratings yet
- 10100Document42 pages10100احمد الدلالNo ratings yet
- International Journal of Heat and Mass TransferDocument11 pagesInternational Journal of Heat and Mass Transferاحمد الدلالNo ratings yet
- Miscellaneous: Sulphur PurificationDocument3 pagesMiscellaneous: Sulphur Purificationاحمد الدلالNo ratings yet
- Standardization of An Economical Bioprocess For Production of Natural Vinegar From SugarcaneDocument7 pagesStandardization of An Economical Bioprocess For Production of Natural Vinegar From Sugarcaneاحمد الدلالNo ratings yet
- Essig EssigfabrikDocument4 pagesEssig Essigfabrikاحمد الدلالNo ratings yet
- Heat Transfer From A Rotating Disk in A Parallel Air CrossflowDocument10 pagesHeat Transfer From A Rotating Disk in A Parallel Air Crossflowاحمد الدلالNo ratings yet
- Petroleum and Petrochemical EngineeringDocument4 pagesPetroleum and Petrochemical Engineeringاحمد الدلالNo ratings yet
- Purification of Sulfur: Raymond F. Bacon Rocco FanelliDocument6 pagesPurification of Sulfur: Raymond F. Bacon Rocco Fanelliاحمد الدلالNo ratings yet
- MecE370 Assignment 4Document1 pageMecE370 Assignment 4احمد الدلالNo ratings yet
- MecE370 Assignment 5Document1 pageMecE370 Assignment 5احمد الدلالNo ratings yet
- United States: Patent OfficeDocument2 pagesUnited States: Patent Officeاحمد الدلالNo ratings yet
- Heat Measurements: Transfer OnDocument11 pagesHeat Measurements: Transfer Onاحمد الدلالNo ratings yet
- Chem3000/Heat Transfer Simulation Assignment-ADocument1 pageChem3000/Heat Transfer Simulation Assignment-Aاحمد الدلالNo ratings yet
- MecE370 Assignment 1Document1 pageMecE370 Assignment 1احمد الدلالNo ratings yet
- Unit Operations of Chemical Engineering: (7th Edition)Document5 pagesUnit Operations of Chemical Engineering: (7th Edition)HennessysNo ratings yet
- Asco CC T LL Coalescer Cartridges 035A CCT 22 UK 7Document2 pagesAsco CC T LL Coalescer Cartridges 035A CCT 22 UK 7Masoud AmirzadehfardNo ratings yet
- Absorption in SemiconductorsDocument24 pagesAbsorption in Semiconductorssanganandan100% (2)
- Euler& Bernolli EquationDocument25 pagesEuler& Bernolli EquationDeyaa MuhammadNo ratings yet
- SACDocument3 pagesSACthehoang12310No ratings yet
- HPHT Intern ReportDocument40 pagesHPHT Intern ReportSURYA VENKATA SAINADH50% (2)
- Nanowski - The Influence of Incondensible Gases PDFDocument6 pagesNanowski - The Influence of Incondensible Gases PDFCaroline SantanaNo ratings yet
- Agar50series MPFM SpecDocument4 pagesAgar50series MPFM SpecJADNo ratings yet
- Selection of Extraction SolventsDocument3 pagesSelection of Extraction SolventsSyafiqah AfandiNo ratings yet
- Test 2 Jj512 Pneumatic and Hydraulic Answers All Question in Part A and Part BDocument5 pagesTest 2 Jj512 Pneumatic and Hydraulic Answers All Question in Part A and Part BZuhaila MohammadNo ratings yet
- 8-9 Sinf Masalalar YechimiDocument223 pages8-9 Sinf Masalalar Yechimiboyc70764100% (1)
- Teodoro, J.K. (HOME EXPERIMENT 1)Document9 pagesTeodoro, J.K. (HOME EXPERIMENT 1)Jherby TeodoroNo ratings yet
- BİOSENSOR MakaleDocument11 pagesBİOSENSOR MakaleTubaNo ratings yet
- The Principles of Ultrasound and Its Application IDocument10 pagesThe Principles of Ultrasound and Its Application ICao Sang NguyễnNo ratings yet
- Amine PKGDocument18 pagesAmine PKGRathinavel PerumalNo ratings yet
- LESSON 4-Solidification of Metals and AlloysDocument19 pagesLESSON 4-Solidification of Metals and Alloysmichael-education KNo ratings yet
- Midsem Exam Question Paper: Semester: 3 Subject: Fluid Flow Operation (3130502)Document2 pagesMidsem Exam Question Paper: Semester: 3 Subject: Fluid Flow Operation (3130502)Hitesh PariharNo ratings yet
- Carburation in Theory Practice A Manual of Reference For Automobile Engineers Owners 1913Document306 pagesCarburation in Theory Practice A Manual of Reference For Automobile Engineers Owners 1913Juan CalfulcuraNo ratings yet
- Lab 1Document3 pagesLab 1milek mathewNo ratings yet
- Book LayoutDocument8 pagesBook LayoutaamenaNo ratings yet
- How Does Gas Injection WorkDocument3 pagesHow Does Gas Injection WorkrajasekharboNo ratings yet
- Gas Lift Valve InjDocument3 pagesGas Lift Valve InjMikhaelrams RamsNo ratings yet
- PUMPS CompressorsLecture NotesDocument16 pagesPUMPS CompressorsLecture NotesAura Paige Montecastro-RevillaNo ratings yet
- George Bridgman Constructive AnatomyDocument18 pagesGeorge Bridgman Constructive AnatomyJishinNo ratings yet
- Chapter 06 Phase Equilibria 4 PDF FreeDocument77 pagesChapter 06 Phase Equilibria 4 PDF FreeGabriel SilvaNo ratings yet
- Mangalore Institute of Technology & Engineering, MoodabidriDocument2 pagesMangalore Institute of Technology & Engineering, MoodabidriNaveen NaviNo ratings yet
- Hall Yarborough ZDocument3 pagesHall Yarborough ZicaNo ratings yet
- Centrifugal CompressorsDocument48 pagesCentrifugal CompressorsSyedahmedkabir IjazfazilNo ratings yet
- L-545 Servicemans ManualDocument56 pagesL-545 Servicemans ManualingenerproNo ratings yet
- Hitachi Magnet HandbookDocument39 pagesHitachi Magnet HandbookSilverio AcuñaNo ratings yet