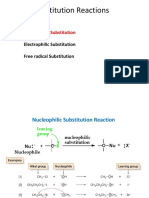
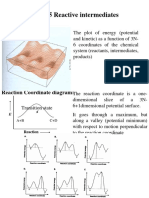
You might also like
- Reagent Function Notes: Any/all 2° R-LDocument10 pagesReagent Function Notes: Any/all 2° R-Lbluebeary22No ratings yet
- Reagen T and Reactio N Mecha Nisms: Physic Organic ChemistryDocument34 pagesReagen T and Reactio N Mecha Nisms: Physic Organic ChemistryUun Khairunnisah KotoNo ratings yet
- UNIT 3-Organic Reactions My VersionDocument47 pagesUNIT 3-Organic Reactions My VersionMohammad JunaidNo ratings yet
- Arrow MechanismDocument49 pagesArrow MechanismtakomolyentinNo ratings yet
- Chemistry-I (For Odd SEMESTER), CY-101: By: Prof. Vandana SrivastavaDocument59 pagesChemistry-I (For Odd SEMESTER), CY-101: By: Prof. Vandana SrivastavaHoly GrailNo ratings yet
- Organicreactionmechanism 160527094347Document55 pagesOrganicreactionmechanism 160527094347Shreyas BhandaryNo ratings yet
- Nucleophilic Substitution ReactionDocument17 pagesNucleophilic Substitution ReactionRojo JohnNo ratings yet
- The Pinacol-Pinacolone RearrangementDocument9 pagesThe Pinacol-Pinacolone RearrangementParag MehtaNo ratings yet
- DR Rafiq Zakaria Campus, Maulana Azad College, DR Ahmad Zaheer Lecture SeriesDocument97 pagesDR Rafiq Zakaria Campus, Maulana Azad College, DR Ahmad Zaheer Lecture Seriesmohsin abdul azizNo ratings yet
- Reaction Mechanisms: Organic ChemistryDocument76 pagesReaction Mechanisms: Organic ChemistryKhenan James NarismaNo ratings yet
- Halo Al KanesDocument17 pagesHalo Al KanesLucas “Khumalo” KaunduNo ratings yet
- Chapter 4 SUBSTITUTION REACTIONDocument35 pagesChapter 4 SUBSTITUTION REACTIONHalimatun MustafaNo ratings yet
- Burrows3e Solutions Ch20Document45 pagesBurrows3e Solutions Ch20Nguyen Duong HieuNo ratings yet
- 4 Alkil Halida PDFDocument39 pages4 Alkil Halida PDFifaNo ratings yet
- Chapter 5 Elimination Reaction - 2016Document19 pagesChapter 5 Elimination Reaction - 2016Syuhadah NoordinNo ratings yet
- 2016 Lect6a Substitution Elimination (Alkilhalides)Document144 pages2016 Lect6a Substitution Elimination (Alkilhalides)Bryan SuryapranataNo ratings yet
- L.S.T. Leung Chik Wai Memorial School F.6 Chemistry Chapter 33: Haloalkane 鹵代化合物 Chpt. 33 p.1Document17 pagesL.S.T. Leung Chik Wai Memorial School F.6 Chemistry Chapter 33: Haloalkane 鹵代化合物 Chpt. 33 p.1Lisa DentonNo ratings yet
- Nucleophilic Substitution An D Elimination ReactionsDocument25 pagesNucleophilic Substitution An D Elimination ReactionsWai Kwong ChiuNo ratings yet
- Alkyl Halides PDFDocument8 pagesAlkyl Halides PDFYSOBELLATHERESE BILOLONo ratings yet
- Substitutions: S 1, S 2: Organic Chemistry (CHEM311) Fall 2005 Dr. Robert F. DiasDocument8 pagesSubstitutions: S 1, S 2: Organic Chemistry (CHEM311) Fall 2005 Dr. Robert F. DiasFan LilyNo ratings yet
- Substitution Elimination (Alkilhalides) Bab 5 FessendenDocument113 pagesSubstitution Elimination (Alkilhalides) Bab 5 Fessendenahmad jamalNo ratings yet
- l4 Alkyl HalidesDocument50 pagesl4 Alkyl HalidesSiti Fatimah0% (1)
- Synthesis of Heterocyclic CompoudsDocument13 pagesSynthesis of Heterocyclic CompoudsYaqeen Alhaqq F. GhaziNo ratings yet
- Synthesis of Heterocyclic CompoudsDocument13 pagesSynthesis of Heterocyclic CompoudsYaqeen Alhaqq F. GhaziNo ratings yet
- Synthesis of Heterocyclic CompoudsDocument13 pagesSynthesis of Heterocyclic CompoudsYaqeen Alhaqq F. GhaziNo ratings yet
- Synthesis of Heterocyclic CompoudsDocument13 pagesSynthesis of Heterocyclic CompoudsYaqeen Alhaqq F. GhaziNo ratings yet
- Alkil HalidaDocument39 pagesAlkil HalidaEva NurfuadaNo ratings yet
- Stereochirality R or SDocument52 pagesStereochirality R or SnifafaniNo ratings yet
- Group 1 - SN1Document11 pagesGroup 1 - SN1faridasaptianiNo ratings yet
- Lecture 32 - TC - 10.11.23Document48 pagesLecture 32 - TC - 10.11.23yakkalivivekNo ratings yet
- Chap. 5 Reactive Intermediates: Energy SurfaceDocument20 pagesChap. 5 Reactive Intermediates: Energy SurfaceAnil KumarNo ratings yet
- Reactivity and Mechanism: Presented By: Dr. Faisal Almalki (Male)Document30 pagesReactivity and Mechanism: Presented By: Dr. Faisal Almalki (Male)battal eduNo ratings yet
- Chapter 6 NotesDocument21 pagesChapter 6 NotesJesús Adrián Gómez OrtizNo ratings yet
- Alkenes - 4Document49 pagesAlkenes - 4Bag CookNo ratings yet
- Stereochemistry of SN Reactions PPT - Copy - Copy-1Document28 pagesStereochemistry of SN Reactions PPT - Copy - Copy-1Vidya Rani100% (2)
- chm207 chp5Document92 pageschm207 chp5Muhd Mirza HizamiNo ratings yet
- Chapter 11Document17 pagesChapter 11abubakarabubakarbah563No ratings yet
- Adisi AlkeneDocument15 pagesAdisi Alkenekurniatriwijaya.2410No ratings yet
- Organic Chemistry Exercise PDFDocument34 pagesOrganic Chemistry Exercise PDFBrightMoonNo ratings yet
- Organic Reaction MechanismsDocument47 pagesOrganic Reaction Mechanismschiomamoronu08No ratings yet
- CH - CH - BR 1° Halide 2° Halide 3° Halide: H H C CH CL CHDocument8 pagesCH - CH - BR 1° Halide 2° Halide 3° Halide: H H C CH CL CHShahbaz NazirNo ratings yet
- Alkenes Reactions NotesDocument14 pagesAlkenes Reactions NotesMartin AlvinNo ratings yet
- Chapter 6 Nucleophilic Substitution On Saturated CarbonsDocument46 pagesChapter 6 Nucleophilic Substitution On Saturated CarbonsNUR HAZWANI BINTI MOHAMAD SANI / UPMNo ratings yet
- RX Adisi Dan SubsitusiDocument48 pagesRX Adisi Dan Subsitusistifar S1A016No ratings yet
- 01 1352193505 80382 PDFDocument86 pages01 1352193505 80382 PDFJennifer Carolina Rosales NoriegaNo ratings yet
- 4 Alkil HalidaDocument39 pages4 Alkil HalidaMuhammad IqbalNo ratings yet
- DR R D Shah 2Document43 pagesDR R D Shah 2yur fanNo ratings yet
- Alkenes - 4Document49 pagesAlkenes - 4Bag CookNo ratings yet
- Photo ChemistryDocument26 pagesPhoto ChemistryAkowuah SamuelNo ratings yet
- Lecture 1 - RecapDocument40 pagesLecture 1 - RecapGemarajuSreeTejaSimhaNo ratings yet
- Free Radicals: OO ONO Molecular Oxygen Nitrogen Dioxide NO Nitrogen MonoxideDocument11 pagesFree Radicals: OO ONO Molecular Oxygen Nitrogen Dioxide NO Nitrogen MonoxideAbdullahi abdulsalamNo ratings yet
- Organic ReactionsDocument50 pagesOrganic ReactionsRyea Chayankka TwentysevendNo ratings yet
- Addition Reactions of AlkynesDocument26 pagesAddition Reactions of Alkynesaggelisgeorge8546No ratings yet
- Ch11 Self-Study PDFDocument22 pagesCh11 Self-Study PDFRida Naila MangiNo ratings yet
- E1 and E2 ReactionsDocument30 pagesE1 and E2 ReactionsVidhu Pandey100% (1)
- Basics of Photochemistry and Norrish Type I Reaction: Presented By: Dr. Nidhi VashisthaDocument12 pagesBasics of Photochemistry and Norrish Type I Reaction: Presented By: Dr. Nidhi Vashisthanidhi vashisthaNo ratings yet
- OrganicChemistryChapter5 PDFDocument19 pagesOrganicChemistryChapter5 PDFJuliet Tatiana CumbeNo ratings yet
- Matriculation Chemistry (Haloalkane)Document46 pagesMatriculation Chemistry (Haloalkane)ridwanNo ratings yet
- Reactions of Ether: Do Not ReactDocument10 pagesReactions of Ether: Do Not ReactcikguhafidzuddinNo ratings yet
- Organometallic Chemistry: Plenary Lectures Presented at the Fourth International Conference on Organometallic ChemistryFrom EverandOrganometallic Chemistry: Plenary Lectures Presented at the Fourth International Conference on Organometallic ChemistryF. G. A. StoneNo ratings yet
- Exp.06 Preparation of P-Bromo AcetanilideDocument3 pagesExp.06 Preparation of P-Bromo AcetanilideAnanda VijayasarathyNo ratings yet
- Organic ChemistryDocument20 pagesOrganic ChemistryGirish RaguvirNo ratings yet
- Har 001-059Document59 pagesHar 001-059yaswanthNo ratings yet
- 2nd Year Chemistry Guess Paper 2022Document6 pages2nd Year Chemistry Guess Paper 2022SaadNo ratings yet
- Cu ZN OrganometallicsDocument60 pagesCu ZN Organometallicsalusia24No ratings yet
- Review Article On Vilsmeier-Haack ReactionDocument19 pagesReview Article On Vilsmeier-Haack Reactionumesh123patil100% (1)
- Carboxylic Acid and Derivatives - JEE Mains PYQ 2020-2022Document74 pagesCarboxylic Acid and Derivatives - JEE Mains PYQ 2020-2022pankaj baidNo ratings yet
- Haloalkanes and Haloarenes Question BankDocument16 pagesHaloalkanes and Haloarenes Question BankBrown HustlerNo ratings yet
- Chapter 15Document43 pagesChapter 15bunnyNo ratings yet
- Organic Chemistry 4th Edition Gorzynski Test BankDocument17 pagesOrganic Chemistry 4th Edition Gorzynski Test Bankdigonousconcrewh2zxi100% (33)
- Subject Chemistry: Paper No and Title 5 Organic Chemistry-II Module No and Title 25 S 1 Reactions Module Tag CHE - P5 - M25Document11 pagesSubject Chemistry: Paper No and Title 5 Organic Chemistry-II Module No and Title 25 S 1 Reactions Module Tag CHE - P5 - M25Kamrujjaman Kamran100% (1)
- Note For All 202L Sections - Spring2014Document82 pagesNote For All 202L Sections - Spring2014yangNo ratings yet
- Halogenalkanes: Unit 2 Chemistry C. Bailey PolackDocument23 pagesHalogenalkanes: Unit 2 Chemistry C. Bailey PolackBritney PattersonNo ratings yet
- 2015 YJC H2 Chem 2015 Prelim Suggested AnswersDocument23 pages2015 YJC H2 Chem 2015 Prelim Suggested AnswerswaimoeNo ratings yet
- AlkanesDocument11 pagesAlkanesДмитрий ЛегаNo ratings yet
- Short Notes Class 12 Chemistry 2023Document37 pagesShort Notes Class 12 Chemistry 2023Susmita BhowmikNo ratings yet
- Hsslive-Xii-Chem-10. Alkyhalides and Aryl HalidesDocument13 pagesHsslive-Xii-Chem-10. Alkyhalides and Aryl HalidesHakim AbbasNo ratings yet
- Halogen Derivatives PDFDocument32 pagesHalogen Derivatives PDFRaju Singh100% (1)
- SN ReactionDocument87 pagesSN ReactionSaptarshi MondalNo ratings yet
- Haloalkanes and Haloarenes: Chapter - 00Document40 pagesHaloalkanes and Haloarenes: Chapter - 00Harsh OthayothNo ratings yet
- Reaction Mechanism Jeemain - GuruDocument12 pagesReaction Mechanism Jeemain - Guruprexa indiaNo ratings yet
- Vivekanandha: Department of Chemistry Master of Science (M.SC.)Document107 pagesVivekanandha: Department of Chemistry Master of Science (M.SC.)Jeevanantham VelayuthamNo ratings yet
- Kimia Part BDocument10 pagesKimia Part Bpkgmuda6869No ratings yet
- Smith Ch08 Lecture EditDocument60 pagesSmith Ch08 Lecture EditfaithNo ratings yet
- CHEM 210 CH 09 Addition ReactionsDocument11 pagesCHEM 210 CH 09 Addition ReactionsJaga SahsinyNo ratings yet
- Iit Questions On Carbonyl Compounds & Carboxylic Acid and Its DerivativeDocument12 pagesIit Questions On Carbonyl Compounds & Carboxylic Acid and Its DerivativeRaju SinghNo ratings yet
- Roman A. Valiulin - Organic Chemistry - 100 Must-Know Mechanisms-Walter de Gruyter (2020)Document250 pagesRoman A. Valiulin - Organic Chemistry - 100 Must-Know Mechanisms-Walter de Gruyter (2020)Daniel Arias100% (1)
- Heterocyclic Compounds Byju FormatDocument112 pagesHeterocyclic Compounds Byju Formatamit tiwariNo ratings yet
- Common Model Exam Set-XIII (B) (2079-3-32) QuestionDocument16 pagesCommon Model Exam Set-XIII (B) (2079-3-32) QuestionSameer KhanNo ratings yet