You might also like
- Genome-Wide Analyses of Exonic Copy Number Variants in A Family-Based Study Point To Novel Autism Susceptibility GenesDocument13 pagesGenome-Wide Analyses of Exonic Copy Number Variants in A Family-Based Study Point To Novel Autism Susceptibility GenesVanessa Llantoy ParraNo ratings yet
- Genome-Wide Association Study Implicates NDST3 in Schizophrenia and Bipolar DisorderDocument10 pagesGenome-Wide Association Study Implicates NDST3 in Schizophrenia and Bipolar Disordera aNo ratings yet
- 10 1002@ajmg B 32750Document8 pages10 1002@ajmg B 32750HeleneNo ratings yet
- ALSDocument14 pagesALSZé CunhaNo ratings yet
- DyslexiahandednessscerriDocument7 pagesDyslexiahandednessscerriapi-302824697No ratings yet
- Role For Hedgehog Signaling in Cranial-Suture Development and ObesityDocument9 pagesRole For Hedgehog Signaling in Cranial-Suture Development and ObesityDanny Alvarez FocacciNo ratings yet
- Arumugam Et Al-Clinical Anatomy 2Document10 pagesArumugam Et Al-Clinical Anatomy 2Kavitha RajaNo ratings yet
- Andreassen Et Al - 2013 - Improved Detection of Common Variants Associated With Schizophrenia and BipolarDocument16 pagesAndreassen Et Al - 2013 - Improved Detection of Common Variants Associated With Schizophrenia and Bipolarkeon.arbabi.altNo ratings yet
- 10 1002/ajmg B 30713Document3 pages10 1002/ajmg B 30713Sri HariNo ratings yet
- First HPSE2 Missense Mutation in Urofacial SyndromeDocument5 pagesFirst HPSE2 Missense Mutation in Urofacial SyndromeAmir Akram MasihNo ratings yet
- 10.1007@s12311 020 01167 XDocument5 pages10.1007@s12311 020 01167 XJesser E. BravoNo ratings yet
- A Higher Mutational Burden in Females Supports A Female Protective Model'' in Neurodevelopmental DisordersDocument11 pagesA Higher Mutational Burden in Females Supports A Female Protective Model'' in Neurodevelopmental Disorderscinfer75No ratings yet
- tmp1329 TMPDocument11 pagestmp1329 TMPFrontiersNo ratings yet
- Genetic Susceptibility To Lupus New Insights From Fine MappingDocument13 pagesGenetic Susceptibility To Lupus New Insights From Fine MappingAlberto Carlos Espinosa GaravitoNo ratings yet
- Michael C O'DonovanDocument3 pagesMichael C O'DonovanFahrunnisa NurdinNo ratings yet
- Omnigenic-Theory (Paper 3)Document10 pagesOmnigenic-Theory (Paper 3)DanNo ratings yet
- The Genetics of Type 2 DiabetesDocument28 pagesThe Genetics of Type 2 DiabetesAmalia Elena BulanceaNo ratings yet
- Psiki 2Document7 pagesPsiki 2Winona ProkNo ratings yet
- Familial Spastic Paraparesis: Evaluation of LocusDocument11 pagesFamilial Spastic Paraparesis: Evaluation of LocusBruno MotaNo ratings yet
- The Disruption of Profiling of Serotonergic Neurons, Results in Autism-Related BehaviorsDocument22 pagesThe Disruption of Profiling of Serotonergic Neurons, Results in Autism-Related BehaviorsAngelinni Taglioni StangeNo ratings yet
- Identification and Characterization of Essential Genes in The Human GenomeDocument7 pagesIdentification and Characterization of Essential Genes in The Human Genome戴义宾No ratings yet
- Genetics of Hypertension: The Lack of EvidenceDocument11 pagesGenetics of Hypertension: The Lack of EvidenceInternational Medical PublisherNo ratings yet
- Molecular AutismDocument27 pagesMolecular AutismJules IuliaNo ratings yet
- Genetics and AlzheimersDocument15 pagesGenetics and AlzheimersSex & Gender Women's Health CollaborativeNo ratings yet
- De Novo Mutations in YWHAG Cause Early Onset EpilepsyDocument11 pagesDe Novo Mutations in YWHAG Cause Early Onset EpilepsyGiselle Costa Daniel HonoratoNo ratings yet
- TMP FDE0Document5 pagesTMP FDE0FrontiersNo ratings yet
- Wang Et Al-2018-Autism ResearchDocument7 pagesWang Et Al-2018-Autism ResearchSTELLA LAZARIDOUNo ratings yet
- David G. Wang, Et Al. - Large-Scale Identification, Mapping, and Genotyping of Single-Nucleotide Polymorphisms in The Human GenomeDocument7 pagesDavid G. Wang, Et Al. - Large-Scale Identification, Mapping, and Genotyping of Single-Nucleotide Polymorphisms in The Human GenomeYopghm698No ratings yet
- Exome Sequencing in Multiplex Autism Families Suggests A Major Role For Heterozygous Truncating MutationsDocument7 pagesExome Sequencing in Multiplex Autism Families Suggests A Major Role For Heterozygous Truncating MutationsTrúc LêNo ratings yet
- Meta-Analysis Shows Strong Positive AssociationDocument8 pagesMeta-Analysis Shows Strong Positive AssociationFahrunnisa NurdinNo ratings yet
- Array-CGH Analysis in A Cohort of Phenotypically Well-Characterized Individuals With "Essential" Autism Spectrum DisordersDocument8 pagesArray-CGH Analysis in A Cohort of Phenotypically Well-Characterized Individuals With "Essential" Autism Spectrum DisordersDaniel VásquezNo ratings yet
- Jurnal AutismDocument10 pagesJurnal AutismAby SuryaNo ratings yet
- Pnas 201117029Document6 pagesPnas 201117029Brandi AllenNo ratings yet
- 32Document8 pages32Guhan KANo ratings yet
- The Relationship of Estrogen Receptor-α and -ß Genes with Osteoarthritis of the HandDocument8 pagesThe Relationship of Estrogen Receptor-α and -ß Genes with Osteoarthritis of the HandIkmal ShahromNo ratings yet
- Association of MSX1 and TGFB3 With Nonsyndromic Clefting in HumansDocument12 pagesAssociation of MSX1 and TGFB3 With Nonsyndromic Clefting in HumansbarcimNo ratings yet
- An Autosomal Genomic Screen For Autism 1999Document7 pagesAn Autosomal Genomic Screen For Autism 1999Harshit AmbeshNo ratings yet
- Otolaryngology Head and Neck Surgery 2014 Bittermann 34 9Document6 pagesOtolaryngology Head and Neck Surgery 2014 Bittermann 34 9Hidayat ArifinNo ratings yet
- Riley 2009Document9 pagesRiley 2009Fahrunnisa NurdinNo ratings yet
- Jurnal 3Document11 pagesJurnal 3meldaiskaNo ratings yet
- A Review of The Role of Female Gender in Autism SpectrumDocument20 pagesA Review of The Role of Female Gender in Autism Spectrummaria fernanda avendañoNo ratings yet
- Analysis of A Neuroscience-Related NewDocument4 pagesAnalysis of A Neuroscience-Related NewTianhui XuNo ratings yet
- Family History of Alzheimer's Disease and Cortical Thickness in Patients With DementiaDocument7 pagesFamily History of Alzheimer's Disease and Cortical Thickness in Patients With DementiaMarcos Josue Costa DiasNo ratings yet
- UROGIN - Matrix Metalloproteinase-1 Expression in Women WithDocument7 pagesUROGIN - Matrix Metalloproteinase-1 Expression in Women WithibyudhistiraavNo ratings yet
- GIGYF1 y AutismoDocument47 pagesGIGYF1 y AutismojaenciNo ratings yet
- Interaction Between ERAP1 and HLA-B27 in Ankylosing Spondylitis Implicates Peptide Handling in The Mechanism For HLA-B27 in Disease SusceptibilityDocument10 pagesInteraction Between ERAP1 and HLA-B27 in Ankylosing Spondylitis Implicates Peptide Handling in The Mechanism For HLA-B27 in Disease SusceptibilityexplodecomputerNo ratings yet
- Phenome Risk Classification Enables Phenotypic Imputation and Gene Discovery in Developmental StutteringDocument27 pagesPhenome Risk Classification Enables Phenotypic Imputation and Gene Discovery in Developmental Stuttering一位大神No ratings yet
- 19857-285991-3-PB - Viii Il 10Document11 pages19857-285991-3-PB - Viii Il 10Anonymous KsxN7vIQxbNo ratings yet
- NG 732Document6 pagesNG 732dryNo ratings yet
- 2014, Nilsson AcanDocument9 pages2014, Nilsson AcanEduMaramNo ratings yet
- Human Pathways in Animal Models-5aDocument13 pagesHuman Pathways in Animal Models-5aElvina DhamayantiNo ratings yet
- Abdellaoui Et Al 2019 (Genes SES UK)Document23 pagesAbdellaoui Et Al 2019 (Genes SES UK)slipinaNo ratings yet
- Campbell-2014-Whole-genome SequencDocument12 pagesCampbell-2014-Whole-genome Sequencpriyaseshadri5No ratings yet
- CMT WEGS Nejmoa0908094Document11 pagesCMT WEGS Nejmoa0908094MCuk2606No ratings yet
- Establishing A Natural History of X-Linked Dystonia ParkinsonismDocument14 pagesEstablishing A Natural History of X-Linked Dystonia ParkinsonismSmilingNo ratings yet
- Ann Rheum Dis 2014 Karaderi Annrheumdis 2014 205643 2Document7 pagesAnn Rheum Dis 2014 Karaderi Annrheumdis 2014 205643 2Wahyu Slamet NugrohoNo ratings yet
- Peopling The AmericasDocument3 pagesPeopling The Americasmikey_tipswordNo ratings yet
- Psychiatry Clin Neurosci - 2017 - Waye - Genetics and Epigenetics of Autism A ReviewDocument17 pagesPsychiatry Clin Neurosci - 2017 - Waye - Genetics and Epigenetics of Autism A ReviewKaline DantasNo ratings yet
- Tissue-Speci Fic Multi-Omics Analysis of Atrial Fibrillation: ArticleDocument15 pagesTissue-Speci Fic Multi-Omics Analysis of Atrial Fibrillation: ArticleAnahí TessaNo ratings yet
- Needle Free Injection TechnologyDocument6 pagesNeedle Free Injection TechnologyPuspala ManojkumarNo ratings yet
- The Complete List of Indian GDRsDocument11 pagesThe Complete List of Indian GDRsJigar ShahNo ratings yet
- Section C - Group 04 - Biocon Ltd.Document17 pagesSection C - Group 04 - Biocon Ltd.Rishabh Goyal100% (1)
- A Comparison Between Cactophilicy East Communities Isolated From Cereus Hildmannianus and Praecereus Euchlorus Necrotic CladodesDocument9 pagesA Comparison Between Cactophilicy East Communities Isolated From Cereus Hildmannianus and Praecereus Euchlorus Necrotic CladodesBárbara MendonçaNo ratings yet
- BioTech Professional ResumeDocument2 pagesBioTech Professional Resumerothko75No ratings yet
- Draget & Taylor (2011) Chemical, Physhicas and Biological Properties of AlginatesDocument6 pagesDraget & Taylor (2011) Chemical, Physhicas and Biological Properties of AlginatesDania Andrea Di FilippoNo ratings yet
- Admission Fall2014Document2 pagesAdmission Fall2014wahabmaths0% (1)
- 11 Chapter 1Document58 pages11 Chapter 1AmanNo ratings yet
- Monalisa ManurungDocument13 pagesMonalisa ManurungPradinta BayuNo ratings yet
- DapusDocument4 pagesDapusVirta GiovanniNo ratings yet
- Bergey's Manual of Systematic Bacteriology - Second EditionDocument401 pagesBergey's Manual of Systematic Bacteriology - Second EditionGF Dogminiq Yenifer0% (2)
- GREDocument7 pagesGREBoris ShalomovNo ratings yet
- Resume Biology SyafaDocument3 pagesResume Biology SyafaSyafa ArumNo ratings yet
- Teva PharmaceuticalsDocument10 pagesTeva PharmaceuticalsivanababicNo ratings yet
- 304 Graphic Organizer AssignmentDocument6 pages304 Graphic Organizer Assignmentapi-376822731No ratings yet
- Httpbarbianatutorial.blogspot.comDocument180 pagesHttpbarbianatutorial.blogspot.comsksarkar.barbianaNo ratings yet
- Laboratory Manual and GuidelineDocument27 pagesLaboratory Manual and GuidelineFatih Rushdi100% (1)
- Bacillus Enzyme Industrial ProductionDocument17 pagesBacillus Enzyme Industrial ProductionBagas Zaki MNo ratings yet
- Sciencemag 28 1 2011Document128 pagesSciencemag 28 1 2011Vika AyuDeviantiNo ratings yet
- Functional Matrix HypothesisDocument16 pagesFunctional Matrix HypothesisUddyot ChaudharyNo ratings yet
- Site-Directed Mutagenesis - Wikipedia, The Free EncyclopediaDocument6 pagesSite-Directed Mutagenesis - Wikipedia, The Free EncyclopediaAlviro CossemeNo ratings yet
- Daku SurajDocument60 pagesDaku SurajDeepak KesharwaniNo ratings yet
- Jurnal Padi PDFDocument7 pagesJurnal Padi PDFGrassellaNo ratings yet
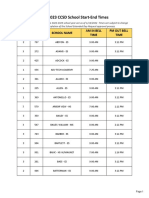
- 2022-2023 CCSD School Start-End TimesDocument23 pages2022-2023 CCSD School Start-End TimesFOX5 Vegas50% (2)
- NIT WARANGAL PhdbrochureDocument17 pagesNIT WARANGAL Phdbrochurevss swamyNo ratings yet
- Jitendra ResumeDocument2 pagesJitendra ResumejitubatistaNo ratings yet
- Genetically Modified OrganismDocument11 pagesGenetically Modified OrganismGigi ArboledaNo ratings yet
- IM Presentation-PRICING STRATEGY (Revised)Document33 pagesIM Presentation-PRICING STRATEGY (Revised)palakNo ratings yet
- Monteiro Et Al., 2003 PDFDocument10 pagesMonteiro Et Al., 2003 PDFGutierrez Guarnizo SneiderNo ratings yet
- New Microsoft Word Document (2Document2 pagesNew Microsoft Word Document (2Shahil AlamNo ratings yet
- When the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisFrom EverandWhen the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisRating: 3.5 out of 5 stars3.5/5 (2)
- 10% Human: How Your Body's Microbes Hold the Key to Health and HappinessFrom Everand10% Human: How Your Body's Microbes Hold the Key to Health and HappinessRating: 4 out of 5 stars4/5 (33)
- The Ancestor's Tale: A Pilgrimage to the Dawn of EvolutionFrom EverandThe Ancestor's Tale: A Pilgrimage to the Dawn of EvolutionRating: 4 out of 5 stars4/5 (811)
- A Brief History of Intelligence: Evolution, AI, and the Five Breakthroughs That Made Our BrainsFrom EverandA Brief History of Intelligence: Evolution, AI, and the Five Breakthroughs That Made Our BrainsRating: 4 out of 5 stars4/5 (5)
- Tales from Both Sides of the Brain: A Life in NeuroscienceFrom EverandTales from Both Sides of the Brain: A Life in NeuroscienceRating: 3 out of 5 stars3/5 (18)
- Gut: the new and revised Sunday Times bestsellerFrom EverandGut: the new and revised Sunday Times bestsellerRating: 4 out of 5 stars4/5 (392)
- Why We Die: The New Science of Aging and the Quest for ImmortalityFrom EverandWhy We Die: The New Science of Aging and the Quest for ImmortalityRating: 4 out of 5 stars4/5 (3)
- Gut: The Inside Story of Our Body's Most Underrated Organ (Revised Edition)From EverandGut: The Inside Story of Our Body's Most Underrated Organ (Revised Edition)Rating: 4 out of 5 stars4/5 (378)
- The Other Side of Normal: How Biology Is Providing the Clues to Unlock the Secrets of Normal and Abnormal BehaviorFrom EverandThe Other Side of Normal: How Biology Is Providing the Clues to Unlock the Secrets of Normal and Abnormal BehaviorNo ratings yet
- The Molecule of More: How a Single Chemical in Your Brain Drives Love, Sex, and Creativity--and Will Determine the Fate of the Human RaceFrom EverandThe Molecule of More: How a Single Chemical in Your Brain Drives Love, Sex, and Creativity--and Will Determine the Fate of the Human RaceRating: 4.5 out of 5 stars4.5/5 (516)
- Undeniable: How Biology Confirms Our Intuition That Life Is DesignedFrom EverandUndeniable: How Biology Confirms Our Intuition That Life Is DesignedRating: 4 out of 5 stars4/5 (11)
- A Series of Fortunate Events: Chance and the Making of the Planet, Life, and YouFrom EverandA Series of Fortunate Events: Chance and the Making of the Planet, Life, and YouRating: 4.5 out of 5 stars4.5/5 (62)
- All That Remains: A Renowned Forensic Scientist on Death, Mortality, and Solving CrimesFrom EverandAll That Remains: A Renowned Forensic Scientist on Death, Mortality, and Solving CrimesRating: 4.5 out of 5 stars4.5/5 (397)
- Good Without God: What a Billion Nonreligious People Do BelieveFrom EverandGood Without God: What a Billion Nonreligious People Do BelieveRating: 4 out of 5 stars4/5 (66)
- Buddha's Brain: The Practical Neuroscience of Happiness, Love & WisdomFrom EverandBuddha's Brain: The Practical Neuroscience of Happiness, Love & WisdomRating: 4 out of 5 stars4/5 (215)
- This Is Your Brain On Parasites: How Tiny Creatures Manipulate Our Behavior and Shape SocietyFrom EverandThis Is Your Brain On Parasites: How Tiny Creatures Manipulate Our Behavior and Shape SocietyRating: 3.5 out of 5 stars3.5/5 (31)
- Fast Asleep: Improve Brain Function, Lose Weight, Boost Your Mood, Reduce Stress, and Become a Better SleeperFrom EverandFast Asleep: Improve Brain Function, Lose Weight, Boost Your Mood, Reduce Stress, and Become a Better SleeperRating: 4.5 out of 5 stars4.5/5 (15)
- Who's in Charge?: Free Will and the Science of the BrainFrom EverandWho's in Charge?: Free Will and the Science of the BrainRating: 4 out of 5 stars4/5 (65)
- The Consciousness Instinct: Unraveling the Mystery of How the Brain Makes the MindFrom EverandThe Consciousness Instinct: Unraveling the Mystery of How the Brain Makes the MindRating: 4.5 out of 5 stars4.5/5 (93)
- The Invention of Tomorrow: A Natural History of ForesightFrom EverandThe Invention of Tomorrow: A Natural History of ForesightRating: 4.5 out of 5 stars4.5/5 (5)
- Crypt: Life, Death and Disease in the Middle Ages and BeyondFrom EverandCrypt: Life, Death and Disease in the Middle Ages and BeyondRating: 4 out of 5 stars4/5 (4)
- The Lives of Bees: The Untold Story of the Honey Bee in the WildFrom EverandThe Lives of Bees: The Untold Story of the Honey Bee in the WildRating: 4.5 out of 5 stars4.5/5 (44)
- Inside of a Dog: What Dogs See, Smell, and KnowFrom EverandInside of a Dog: What Dogs See, Smell, and KnowRating: 4 out of 5 stars4/5 (390)
- Wayfinding: The Science and Mystery of How Humans Navigate the WorldFrom EverandWayfinding: The Science and Mystery of How Humans Navigate the WorldRating: 4.5 out of 5 stars4.5/5 (18)