You might also like
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- Weather Modification CL 2009Document9 pagesWeather Modification CL 2009javenacostaNo ratings yet
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Respuesta Geofisica de La Ionosfera A HaarpDocument5 pagesRespuesta Geofisica de La Ionosfera A HaarpjavenacostaNo ratings yet
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Bio-Based Levulinic Acid, Furfural, and Hydroxymethylfurfural in WaterDocument13 pagesBio-Based Levulinic Acid, Furfural, and Hydroxymethylfurfural in WaterjavenacostaNo ratings yet
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- StressDocument1 pageStressjavenacostaNo ratings yet
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Innan Estimulando Ondas en La Magnetosfera Con HaarpDocument2 pagesInnan Estimulando Ondas en La Magnetosfera Con HaarpjavenacostaNo ratings yet
- Entrenamiento de EEUU A Rebeldes SiriosDocument44 pagesEntrenamiento de EEUU A Rebeldes SiriosjavenacostaNo ratings yet
- Critical HF Heating Power For Major Enhancement of Elf Wave. Generation by Modulated ElectrojetDocument4 pagesCritical HF Heating Power For Major Enhancement of Elf Wave. Generation by Modulated ElectrojetjavenacostaNo ratings yet
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- 2016 DTRA ContractorsDocument10 pages2016 DTRA ContractorsjavenacostaNo ratings yet
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- 03 Neem Turmeric in Treatment of Covid 19Document2 pages03 Neem Turmeric in Treatment of Covid 19javenacostaNo ratings yet
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- COVID-19 Pandemic Is A War Crime Against HumanityDocument41 pagesCOVID-19 Pandemic Is A War Crime Against HumanityjavenacostaNo ratings yet
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- SARS-CoV-2 Mass Vaccination - Urgent Questions On Vaccine SafetyDocument10 pagesSARS-CoV-2 Mass Vaccination - Urgent Questions On Vaccine SafetyjavenacostaNo ratings yet
- The Lab-Leak HypothesisDocument32 pagesThe Lab-Leak Hypothesisjavenacosta100% (1)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Boyle, F. A - SARS-cov-2 Is A Biological Warfare WeaponDocument9 pagesBoyle, F. A - SARS-cov-2 Is A Biological Warfare Weaponjavenacosta100% (1)
- Bats As Reservoir For SARSDocument4 pagesBats As Reservoir For SARSjavenacostaNo ratings yet
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- Covid Vaccine Mortality PDFDocument11 pagesCovid Vaccine Mortality PDFjavenacosta100% (4)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- Prediction and Prevention of The Next Pandemic ZoonosisDocument10 pagesPrediction and Prevention of The Next Pandemic ZoonosisjavenacostaNo ratings yet
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- Daszak Et Al - EID - Science 2000Document7 pagesDaszak Et Al - EID - Science 2000javenacostaNo ratings yet
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Covid-19 Bio-Warfare and The 21ST Century PandemicDocument9 pagesCovid-19 Bio-Warfare and The 21ST Century PandemicjavenacostaNo ratings yet
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Simulacion de Fakes News en EpidemiasDocument10 pagesSimulacion de Fakes News en EpidemiasjavenacostaNo ratings yet
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
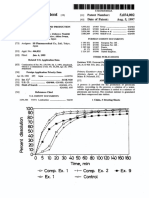
- +-Comp. Ex. 2 - Ex. 9: United States PatentDocument9 pages+-Comp. Ex. 2 - Ex. 9: United States PatentjavenacostaNo ratings yet
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- 7 HMP Shunt Pathway RO PDF v1Document9 pages7 HMP Shunt Pathway RO PDF v1Radwa MohamedNo ratings yet
- Diabetes MellitusDocument24 pagesDiabetes MellitusIgwe SolomonNo ratings yet
- Daksh MahajanDocument12 pagesDaksh MahajanslippypizzaNo ratings yet
- Gene Expression Literature ReviewDocument7 pagesGene Expression Literature Reviewafdtyaxfv100% (1)
- Article WJPR 1538203814 PDFDocument13 pagesArticle WJPR 1538203814 PDFAnant SrivastavaNo ratings yet
- DLL SHS STEM Science Grade 11 - Earth & Life Quarter2 Week4 (Palawan Division)Document14 pagesDLL SHS STEM Science Grade 11 - Earth & Life Quarter2 Week4 (Palawan Division)Maylene VillarosaNo ratings yet
- Biologia Telomerilor Umani2711Document20 pagesBiologia Telomerilor Umani2711Somnis VeritasNo ratings yet
- Act04 Cell Division - Mitosis DiscussionDocument3 pagesAct04 Cell Division - Mitosis DiscussionPaula Nicole AlanoNo ratings yet
- PHA611 LEC 1st ShiftingDocument225 pagesPHA611 LEC 1st ShiftingGAILE MEIZTY MOSADANo ratings yet
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Reaksi Terang Dan Gelap: Dua Tahapan Fotosintesis Tanaman Sekar Arum Wulandari (1302619067)Document7 pagesReaksi Terang Dan Gelap: Dua Tahapan Fotosintesis Tanaman Sekar Arum Wulandari (1302619067)Adila KurniatiNo ratings yet
- VDJ Recombination Machanism InitiationDocument38 pagesVDJ Recombination Machanism InitiationBogdan HateganNo ratings yet
- Lecture 5Document26 pagesLecture 5BlaNo ratings yet
- Integration of MetabolismDocument44 pagesIntegration of MetabolismHalidaaNo ratings yet
- Regulation of Gene ExpressionDocument34 pagesRegulation of Gene ExpressionJay Fickle100% (1)
- Mitochondria: Structure and FunctionDocument6 pagesMitochondria: Structure and FunctionHariniNo ratings yet
- Science: Mahay Integrated Secondary SchoolDocument8 pagesScience: Mahay Integrated Secondary Schoollavenia acdalNo ratings yet
- AP Biology Free Response On Protein SynthesisDocument7 pagesAP Biology Free Response On Protein Synthesisafktgvllsgajfw100% (2)
- DNA Puzzle Kit: Teacher'S Manual and Student GuideDocument3 pagesDNA Puzzle Kit: Teacher'S Manual and Student GuideClven Fernandez0% (1)
- DNA, RNA & Protein SynthesisDocument21 pagesDNA, RNA & Protein Synthesiscmillica1176100% (3)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Cell Bio Exam 1 Lecture QuizzesDocument50 pagesCell Bio Exam 1 Lecture QuizzesHafiza Dia Islam100% (1)
- Downloadable Test Bank For Fundamentals of Human Physiology 4th Edition Sherwood FPHP4e - 002TB 1Document22 pagesDownloadable Test Bank For Fundamentals of Human Physiology 4th Edition Sherwood FPHP4e - 002TB 1Zaid Khaled100% (1)
- Bab.4. AnabolismeDocument14 pagesBab.4. AnabolismeSurniSajaNo ratings yet
- Honors+Onion+Root+Mitosis+Lab VirtualDocument5 pagesHonors+Onion+Root+Mitosis+Lab VirtualSwarra MudgalkarNo ratings yet
- HM 11Document51 pagesHM 11body fayezNo ratings yet
- 11 - Carbohydrate MetabolismDocument68 pages11 - Carbohydrate MetabolismcheckmateNo ratings yet
- Comparing and Contrasting C3, C4, CAM - AP BiologyDocument1 pageComparing and Contrasting C3, C4, CAM - AP BiologyFVCproductionsNo ratings yet
- Amino Acid MetabolismDocument7 pagesAmino Acid MetabolismROHITNo ratings yet
- A Seminar Presentation On Mechanism of Drug ActionDocument40 pagesA Seminar Presentation On Mechanism of Drug Actionbellatrix aliaNo ratings yet
- Biochemistry Lecture ScheduleDocument1 pageBiochemistry Lecture ScheduleTheBoss 20No ratings yet
- 18 Cell Cycle Regulation-SDocument6 pages18 Cell Cycle Regulation-SkriishnaNo ratings yet
- Return of the God Hypothesis: Three Scientific Discoveries That Reveal the Mind Behind the UniverseFrom EverandReturn of the God Hypothesis: Three Scientific Discoveries That Reveal the Mind Behind the UniverseRating: 4.5 out of 5 stars4.5/5 (52)
- The Rise and Fall of the Dinosaurs: A New History of a Lost WorldFrom EverandThe Rise and Fall of the Dinosaurs: A New History of a Lost WorldRating: 4 out of 5 stars4/5 (597)
- Why We Die: The New Science of Aging and the Quest for ImmortalityFrom EverandWhy We Die: The New Science of Aging and the Quest for ImmortalityRating: 4 out of 5 stars4/5 (5)
- A Brief History of Intelligence: Evolution, AI, and the Five Breakthroughs That Made Our BrainsFrom EverandA Brief History of Intelligence: Evolution, AI, and the Five Breakthroughs That Made Our BrainsRating: 4.5 out of 5 stars4.5/5 (6)