You might also like
- Vries-Driessen2011 Article EducationalPaperDocument9 pagesVries-Driessen2011 Article EducationalPaperCristian HaroNo ratings yet
- Cvid Lancet 2008Document14 pagesCvid Lancet 2008Andre GarciaNo ratings yet
- Jurnal 2Document8 pagesJurnal 2tasya tazkia regita zahraNo ratings yet
- Immunode Ficiency Disorders: Education GapsDocument16 pagesImmunode Ficiency Disorders: Education GapsNadejda MarcovaNo ratings yet
- Overview of Immunodeficiency DisordersDocument25 pagesOverview of Immunodeficiency DisordersLuisa AlandeteNo ratings yet
- Immunodeficiency BriefingDocument4 pagesImmunodeficiency BriefingAldiyanzah Lukman100% (1)
- A Rose Is A Rose Is A Rose But CVID Is Not CVID - Common Variable Immune Deficiency (CVID) What Do We Know in 2011Document61 pagesA Rose Is A Rose Is A Rose But CVID Is Not CVID - Common Variable Immune Deficiency (CVID) What Do We Know in 2011vishnupgiNo ratings yet
- Materi Dr. R. A. Myrna Alia, M.kes, Sp.a (K)Document33 pagesMateri Dr. R. A. Myrna Alia, M.kes, Sp.a (K)Handoyo KooNo ratings yet
- Ten Warning Signs of Primary Immunodeficiency: A New Paradigm Is Needed For The 21st CenturyDocument8 pagesTen Warning Signs of Primary Immunodeficiency: A New Paradigm Is Needed For The 21st CenturyVette Angelikka Dela CruzNo ratings yet
- Primary Immunodeficiency: Review Open AccessDocument8 pagesPrimary Immunodeficiency: Review Open AccessMutiara ChairunnisaNo ratings yet
- IPOPI Booklet VaccinesDocument12 pagesIPOPI Booklet VaccinesnarNo ratings yet
- Pproach To The Child With Recurrent Infections Presentation and Investigation of Primary ImmunodeficiencyDocument5 pagesPproach To The Child With Recurrent Infections Presentation and Investigation of Primary ImmunodeficiencyTri Ayu WdNo ratings yet
- Autoimmunity in Common Variable ImmunodeficiencyDocument11 pagesAutoimmunity in Common Variable ImmunodeficiencyAldito GlasgowNo ratings yet
- Primary Immunodeficiency - PMCDocument1 pagePrimary Immunodeficiency - PMCfaniNo ratings yet
- Common Variable Immunodeficiency: Epidemiology, Pathogenesis, Clinical Manifestations, Diagnosis, Classification, and ManagementDocument21 pagesCommon Variable Immunodeficiency: Epidemiology, Pathogenesis, Clinical Manifestations, Diagnosis, Classification, and ManagementViviana LunaNo ratings yet
- Ijms 21 05223 PDFDocument42 pagesIjms 21 05223 PDFrohailNo ratings yet
- Cytomegalovirus in Primary ImmunodeficiencyDocument9 pagesCytomegalovirus in Primary ImmunodeficiencyAle Pushoa UlloaNo ratings yet
- Infectious and Noninfectious Pulmonary Complications in Patients With Primary Immunodeficiency DisordersDocument12 pagesInfectious and Noninfectious Pulmonary Complications in Patients With Primary Immunodeficiency Disorderskim leNo ratings yet
- Immune DeficiencyDocument18 pagesImmune DeficiencyDr anas AbdullahNo ratings yet
- Autoinmunidad e InmunodeficienciaDocument9 pagesAutoinmunidad e InmunodeficienciaJorge Verdugo MuñozNo ratings yet
- Szczawinska Poplonyk2009 PDFDocument12 pagesSzczawinska Poplonyk2009 PDFFernando CruzNo ratings yet
- Chronic Mucocutaneous Candidiasis Report of Three Cases With Different PhenotypesDocument4 pagesChronic Mucocutaneous Candidiasis Report of Three Cases With Different PhenotypesDhilah Harfadhilah FakhirahNo ratings yet
- CMV Infection As A Possible Cause of Chronic Diarrhea in Patient With Autosomal Recessive Hyper Ige Syndrome Case ReportDocument2 pagesCMV Infection As A Possible Cause of Chronic Diarrhea in Patient With Autosomal Recessive Hyper Ige Syndrome Case ReportBIOMEDSCIDIRECT PUBLICATIONSNo ratings yet
- ImmunodeficiencyDocument18 pagesImmunodeficiencyjannah.orangeNo ratings yet
- 2448 9190 Ram 67 03 202Document12 pages2448 9190 Ram 67 03 202anonimo exeNo ratings yet
- State-Of-The-Art Diagnostic Evaluation of Common Variable ImmunodeficiencyDocument9 pagesState-Of-The-Art Diagnostic Evaluation of Common Variable ImmunodeficiencyGreen LightNo ratings yet
- Chapter 15 - The Immunocompromised Host - APIC Text OnlineDocument35 pagesChapter 15 - The Immunocompromised Host - APIC Text OnlineManalAbdelazizNo ratings yet
- Amaya Uribe2019Document21 pagesAmaya Uribe2019kim leNo ratings yet
- 07 Intravenous Immunoglobulin Replacement Therapy in Children With Primary Immunodeficiency Diseases A Nurse S GuideDocument9 pages07 Intravenous Immunoglobulin Replacement Therapy in Children With Primary Immunodeficiency Diseases A Nurse S GuidekiminoooNo ratings yet
- Immune Deficincies - SarahDocument10 pagesImmune Deficincies - SarahkhaledNo ratings yet
- Is Lec Immunodeficiency DiseasesDocument9 pagesIs Lec Immunodeficiency DiseasesKez AmolatoNo ratings yet
- Diagnostic_Testing_Procedures_Document6 pagesDiagnostic_Testing_Procedures_Sarah Marie ArquizaNo ratings yet
- Immunodeficiency 2017 FinalA - 304963 - 284 - 2104 - v3Document6 pagesImmunodeficiency 2017 FinalA - 304963 - 284 - 2104 - v3Muhammed Hashim MNo ratings yet
- Reviewofprimary ImmunodeficiencydiseasesDocument58 pagesReviewofprimary Immunodeficiencydiseasespriti adsulNo ratings yet
- Caso Clínico 1Document24 pagesCaso Clínico 1VillafuerteLizGabyNo ratings yet
- 1 s2.0 S2352304219300881 MainDocument12 pages1 s2.0 S2352304219300881 Mainpdfs studiesNo ratings yet
- Cancers Related To Immunodeficiencies: Update and PerspectivesDocument13 pagesCancers Related To Immunodeficiencies: Update and PerspectivesHabiby Habibaty QolbiNo ratings yet
- Pediatric ImmunologyDocument12 pagesPediatric ImmunologyCharles BayogNo ratings yet
- Current Approach To Primary Immunodeficiency Diseases: March 2019Document9 pagesCurrent Approach To Primary Immunodeficiency Diseases: March 2019Ryan ProxyNo ratings yet
- Reviews Mechanisms, biomarkers and targets for adult-onset Still’s diseaseDocument16 pagesReviews Mechanisms, biomarkers and targets for adult-onset Still’s diseaseRafaela PerezNo ratings yet
- Immunodeficiency Diseases: Professor Shahenaz M.HussienDocument17 pagesImmunodeficiency Diseases: Professor Shahenaz M.HussienVINAY KUMAR DHAWANNo ratings yet
- Pediatric HIV Immune Reconstitution Inflammatory Syndrome (IRISDocument15 pagesPediatric HIV Immune Reconstitution Inflammatory Syndrome (IRISFihzanNo ratings yet
- DR - Dinesh AgarwalDocument20 pagesDR - Dinesh AgarwalPushpanjali Crosslay HospitalNo ratings yet
- Babesiosis in Pregnancy: An Imitator of HELLP SyndromeDocument6 pagesBabesiosis in Pregnancy: An Imitator of HELLP Syndromemade dharmaNo ratings yet
- Immunocompromised DefinitionDocument5 pagesImmunocompromised Definitionapi-511284651No ratings yet
- Ijns 07 00074Document13 pagesIjns 07 00074sunilpkumar18No ratings yet
- Pulmonary Manifestation of PIDDocument14 pagesPulmonary Manifestation of PIDYandiNo ratings yet
- IMMUNOLOGY DISORDERS EXPLAINEDDocument27 pagesIMMUNOLOGY DISORDERS EXPLAINEDShadma KhanNo ratings yet
- Nelson 2017Document28 pagesNelson 2017Santiago GonzalezNo ratings yet
- Management of Patients With Immune Deficiency DisordersDocument11 pagesManagement of Patients With Immune Deficiency DisordersmasheennavirgoNo ratings yet
- MLS 704 Presentation #2Document46 pagesMLS 704 Presentation #2NjeodoNo ratings yet
- Pediatric Immunology: A Case-Based Collection with MCQs, Volume 2From EverandPediatric Immunology: A Case-Based Collection with MCQs, Volume 2No ratings yet
- Immunological Changes With Age and Innovative Approaches To Bolster Immune Function in Older AdultsDocument11 pagesImmunological Changes With Age and Innovative Approaches To Bolster Immune Function in Older AdultsKIU PUBLICATION AND EXTENSIONNo ratings yet
- Ni Hms 316844Document15 pagesNi Hms 316844ntnquynhproNo ratings yet
- Seminar: Enno Schmidt, Detlef ZillikensDocument13 pagesSeminar: Enno Schmidt, Detlef ZillikensUssiy RachmanNo ratings yet
- CHAPTER 6 Infection and DiseasesDocument17 pagesCHAPTER 6 Infection and DiseasesDanica DaniotNo ratings yet
- Allergic Diseases in The Elderly: Review Open AccessDocument10 pagesAllergic Diseases in The Elderly: Review Open AccessAngga Aryo LukmantoNo ratings yet
- Peacock Et Al-2016-Oral DiseasesDocument23 pagesPeacock Et Al-2016-Oral DiseasesGgg2046No ratings yet
- Pediatrics - ImmunodeficiencyDocument3 pagesPediatrics - ImmunodeficiencyJasmine KangNo ratings yet
- Manifestation of Aids With Diarhea 1Document6 pagesManifestation of Aids With Diarhea 1nadyaNo ratings yet
- 143-MPhil Biochemistry-1st-1Document2 pages143-MPhil Biochemistry-1st-1Muhammad ShaikhNo ratings yet
- ANALYTICAL ERRORS OF COLLECTING BLOOD FOR CALCIUM TESTINGDocument17 pagesANALYTICAL ERRORS OF COLLECTING BLOOD FOR CALCIUM TESTINGMuhammad ShaikhNo ratings yet
- Bleeding Disorders 082508Document46 pagesBleeding Disorders 082508Muhammad ShaikhNo ratings yet
- Complete Blood Count Examination By:Muhammad Danyal Hayat and Hamza Sarwar Why CBC Is Performed?Document10 pagesComplete Blood Count Examination By:Muhammad Danyal Hayat and Hamza Sarwar Why CBC Is Performed?Muhammad ShaikhNo ratings yet
- Bio SafetyDocument18 pagesBio SafetyMuhammad ShaikhNo ratings yet
- Assessment of Kidney Diseases by Urinalysis: Presented by Group ADocument22 pagesAssessment of Kidney Diseases by Urinalysis: Presented by Group AMuhammad ShaikhNo ratings yet
- NAB Advertisement for Data Entry, Steno, Clerk PositionsDocument5 pagesNAB Advertisement for Data Entry, Steno, Clerk PositionsTanveer Ahmad AfridiNo ratings yet
- NAB Advertisement for Data Entry, Steno, Clerk PositionsDocument5 pagesNAB Advertisement for Data Entry, Steno, Clerk PositionsTanveer Ahmad AfridiNo ratings yet
- ANALYTICAL ERRORS OF COLLECTING BLOOD FOR CALCIUM TESTINGDocument17 pagesANALYTICAL ERRORS OF COLLECTING BLOOD FOR CALCIUM TESTINGMuhammad ShaikhNo ratings yet
- Nelsen ASCPBoardCertificationExamDocument21 pagesNelsen ASCPBoardCertificationExamMuhammad ShaikhNo ratings yet
- Usage History: Total Balance Used Rs 0.0Document4 pagesUsage History: Total Balance Used Rs 0.0Muhammad ShaikhNo ratings yet
- 2Document3 pages2Muhammad ShaikhNo ratings yet
- Urinalysis Sample Report With Notes - 0Document1 pageUrinalysis Sample Report With Notes - 0sukanto bagchiNo ratings yet
- Histotechnology 4e LookInsideDocument51 pagesHistotechnology 4e LookInsideWildercino RA17% (6)
- Medical Lab TechnologyDocument168 pagesMedical Lab Technologyandrei_niculae_2No ratings yet
- LN Urinalysis FinalDocument138 pagesLN Urinalysis FinalMohsen HaleemNo ratings yet
- Diagnosis of Primary ImmunodeficienciesDocument12 pagesDiagnosis of Primary ImmunodeficienciesMuhammad ShaikhNo ratings yet
- Clinical Chemistry - Learning Guide SeriesDocument117 pagesClinical Chemistry - Learning Guide SeriesjonasNo ratings yet
- Jact01i1p39 PDFDocument12 pagesJact01i1p39 PDFMuflih MahsyarNo ratings yet
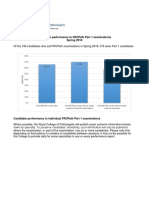
- Examination Performance Report Spring 2018 Part 1Document3 pagesExamination Performance Report Spring 2018 Part 1Syed Danish AliNo ratings yet
- Template For Staff Development PlanDocument1 pageTemplate For Staff Development PlanKenrick BajaoNo ratings yet
- AMS Symptoms and CategoriesDocument1 pageAMS Symptoms and CategoriesyyggvcNo ratings yet
- Victoria DePonceau SPD ResumeDocument1 pageVictoria DePonceau SPD ResumeVictoria Leigh DePonceauNo ratings yet
- Higado Graso No Alcoholico y Niveles de PCRDocument6 pagesHigado Graso No Alcoholico y Niveles de PCRperuliz19No ratings yet
- B. Bruce Myers, MD PHD FACSDocument5 pagesB. Bruce Myers, MD PHD FACSallison8844No ratings yet
- Shih Tzus: What A Unique Breed!Document5 pagesShih Tzus: What A Unique Breed!Jimmy YauNo ratings yet
- Contractor Safety ManualDocument145 pagesContractor Safety Manualdaladar2008No ratings yet
- Abnormal Psychology Beidel 3rd Edition Test BankDocument4 pagesAbnormal Psychology Beidel 3rd Edition Test BankChuck Simmons100% (25)
- COSH Report - Sir KashmerDocument123 pagesCOSH Report - Sir Kashmerengr rangNo ratings yet
- Diabetes Care in Nigeria - 2015 - Annals of Global Health PDFDocument9 pagesDiabetes Care in Nigeria - 2015 - Annals of Global Health PDFarmanNo ratings yet
- The Essentials of Contraceptive TechnologyDocument355 pagesThe Essentials of Contraceptive TechnologyIndah PurnamasariNo ratings yet
- Psychological JournalDocument3 pagesPsychological JournalDiyah CandraNo ratings yet
- Comparison of Supervised Exercise and Home Exercise After Ankle FractureDocument6 pagesComparison of Supervised Exercise and Home Exercise After Ankle FractureKaren MuñozNo ratings yet
- Full Download Test Bank For Health The Basics 10th Edition by Donatelle PDF Full ChapterDocument36 pagesFull Download Test Bank For Health The Basics 10th Edition by Donatelle PDF Full Chapterpedagog.blocage.5iwtr100% (16)
- Mel Zack 1975Document23 pagesMel Zack 1975Mohammad Jofa Rachman PNo ratings yet
- Conduct Disorder Fact Sheet - K. Elise Parker PDFDocument2 pagesConduct Disorder Fact Sheet - K. Elise Parker PDFepaka02No ratings yet
- The Pan StationsDocument14 pagesThe Pan StationsmuradkhanNo ratings yet
- Proposal Presentation Nairobi - Respicius Shumbusho DamianDocument13 pagesProposal Presentation Nairobi - Respicius Shumbusho DamianShumbusho ConsultingtzNo ratings yet
- No Article Title Time of Publication Authors Journal Name Received Date Revised Date Accepted DateDocument4 pagesNo Article Title Time of Publication Authors Journal Name Received Date Revised Date Accepted DateAnin Kalma MartadinataNo ratings yet
- Occupational Health and Safety Issues of Traffic EnforcersDocument16 pagesOccupational Health and Safety Issues of Traffic EnforcersRhobin Mhark GabitoNo ratings yet
- Pediatric Malnutrition Causes and ManagementDocument38 pagesPediatric Malnutrition Causes and ManagementShafie ElmiNo ratings yet
- Table 6-1 Minimum Filter Efficiencies: ANSI /ASHRAE /ASHE Standard 170-2008Document4 pagesTable 6-1 Minimum Filter Efficiencies: ANSI /ASHRAE /ASHE Standard 170-2008Yousef AlalawiNo ratings yet
- Mental Health EssayDocument2 pagesMental Health EssayROHITNo ratings yet
- CPG Gestational Trophoblastic DiseasesDocument31 pagesCPG Gestational Trophoblastic DiseasesSMR50% (2)
- Lecture 4 Safety Engineering PracticesDocument40 pagesLecture 4 Safety Engineering PracticesPaolo Manuel NagayoNo ratings yet
- Diabetes Type 2 BasicsDocument44 pagesDiabetes Type 2 BasicsMasunji EmanuelNo ratings yet
- The Family Doctor - Prof. Henry S Taylor, M.D., 1860Document324 pagesThe Family Doctor - Prof. Henry S Taylor, M.D., 1860IreneRainsNo ratings yet
- Tobacco Free Generation Next - M. Amebari NongsiejDocument2 pagesTobacco Free Generation Next - M. Amebari NongsiejM. Amebari NongsiejNo ratings yet
- Patient Information Guide: AH-QF-FO-07Document35 pagesPatient Information Guide: AH-QF-FO-07jose rajanNo ratings yet