You might also like
- Lipid Analysis: Melisa Intan BarlianaDocument38 pagesLipid Analysis: Melisa Intan BarlianaChantique Maharani0% (1)
- Properties of LipidsDocument12 pagesProperties of Lipidskolita kamal100% (1)
- Estimation of Free Fatty Acids (Acid Values) in Oilsfats by Direct Titration MethodDocument1 pageEstimation of Free Fatty Acids (Acid Values) in Oilsfats by Direct Titration MethodMuhammad Aslam100% (2)
- Oil AnalysisDocument11 pagesOil AnalysisTowfiq Hossain Tasku100% (1)
- EXP 2 RancidityDocument6 pagesEXP 2 RancidityNur SyahirahNo ratings yet
- Peroxide ValueDocument2 pagesPeroxide ValueanthorNo ratings yet
- Biochemistry LaboratoryDocument7 pagesBiochemistry LaboratoryAIra OrtegaNo ratings yet
- Extraction of Total Lipids From Chicken Egg Yolk, Column Chromatography and Qualitative Tests For LipidsDocument10 pagesExtraction of Total Lipids From Chicken Egg Yolk, Column Chromatography and Qualitative Tests For Lipidsmarilujane75% (8)
- Cognis College: Fatty AcidsDocument22 pagesCognis College: Fatty AcidsRidhuan Dion100% (3)
- Determination of AshDocument9 pagesDetermination of Ashkolita kamal100% (4)
- Proximate AnalysisDocument42 pagesProximate AnalysisMonica NCNo ratings yet
- Qualitative Tests For Fats and OilsDocument1 pageQualitative Tests For Fats and OilsMuhammad Aslam100% (1)
- Butter ManufactureDocument14 pagesButter ManufactureKSHETRIMAYUM MONIKA DEVINo ratings yet
- Title: Aim: Date: Name: Matriculation Number: Serial Number: Group: Experiment Number: Level: Course Code: Instructor'S NameDocument14 pagesTitle: Aim: Date: Name: Matriculation Number: Serial Number: Group: Experiment Number: Level: Course Code: Instructor'S NameJim0% (1)
- Lecithin - A Useful Byproduct of Edible Oil ExtractionDocument11 pagesLecithin - A Useful Byproduct of Edible Oil Extractionadda100% (1)
- Qualitative Analysis of Amino Acids and Proteins and Carbohydrates and Lipids and SteroidsDocument35 pagesQualitative Analysis of Amino Acids and Proteins and Carbohydrates and Lipids and SteroidsgangsNo ratings yet
- Qualitative and Quantitative Tests For Lipids.: Prof. Liwayway Memije-CruzDocument11 pagesQualitative and Quantitative Tests For Lipids.: Prof. Liwayway Memije-CruzAurian TormesNo ratings yet
- 1 Determination of Total Acidity of FoodDocument27 pages1 Determination of Total Acidity of FoodVismayNo ratings yet
- Characterization of LipidsDocument4 pagesCharacterization of LipidsEzraela Neriah AmodoNo ratings yet
- Chemistry of Fats & OilsDocument38 pagesChemistry of Fats & OilsSubhajit SarkarNo ratings yet
- Qualitative Analysis of Lipids Through The Extraction of Total Lipids From Chicken Egg YolkDocument5 pagesQualitative Analysis of Lipids Through The Extraction of Total Lipids From Chicken Egg YolkJessa Mateum VallangcaNo ratings yet
- Hydrogenation of Vegetable OilDocument50 pagesHydrogenation of Vegetable OilNoman Aslam100% (2)
- Acrolein Test: Identifying Lipids Using Chemical TestsDocument3 pagesAcrolein Test: Identifying Lipids Using Chemical TestsHenna TkNo ratings yet
- Mayonaise Production ProcessDocument22 pagesMayonaise Production ProcessZulu Alpha100% (2)
- Expt 2 BIOCHEMLABDocument5 pagesExpt 2 BIOCHEMLABEloisah Vin Santiago Ragodon100% (1)
- Quantitative Analysis of Carbohydrates I - Lab 4Document27 pagesQuantitative Analysis of Carbohydrates I - Lab 4Noriko Medoruma0% (3)
- 2.metabolism of LipidsDocument159 pages2.metabolism of LipidsChristian Tham100% (1)
- Isolation, Purification, and Partial Characterization of A Lipid Extract From NutmegDocument12 pagesIsolation, Purification, and Partial Characterization of A Lipid Extract From NutmegJulio Francisco100% (2)
- Protein AnalysisDocument29 pagesProtein AnalysisNaufal Qaweim100% (1)
- Peroxide Value DeterminationDocument5 pagesPeroxide Value DeterminationJohn Paul Pasicaran75% (20)
- Extraction of Total Lipids From Chicken Egg Yolk and Qualitative Test For LipidsDocument4 pagesExtraction of Total Lipids From Chicken Egg Yolk and Qualitative Test For LipidsKizer Dela CruzNo ratings yet
- Alcohols and PhenolsDocument8 pagesAlcohols and PhenolsMomer83% (6)
- Methods of Analysis Processed Fruits and Vegetables, FinalDocument60 pagesMethods of Analysis Processed Fruits and Vegetables, FinalAnkur Bhavsar100% (2)
- Saponification Value of OilDocument16 pagesSaponification Value of OilJim0% (1)
- Crude Fiber Lab Report - Docx Updated FileDocument7 pagesCrude Fiber Lab Report - Docx Updated FileNicholas Boampong100% (2)
- LipidsDocument22 pagesLipidsJoren Angela Galang83% (6)
- Alcohol FermentationDocument34 pagesAlcohol FermentationSaravana Bharathy ReddyNo ratings yet
- Analysis of Lipids in Egg YolkDocument2 pagesAnalysis of Lipids in Egg YolkHydieNo ratings yet
- Lecture 1 Basic Principles of Food Fermentation 1487564123Document37 pagesLecture 1 Basic Principles of Food Fermentation 1487564123Dr. Gurumurthy D M100% (1)
- Classification of EmulsifierDocument17 pagesClassification of EmulsifierAsyraf Anas100% (4)
- Ex6 Peroxide ValueDocument7 pagesEx6 Peroxide ValueChidi IfenweobiNo ratings yet
- Experiment 9 Total Serum CholesterolDocument8 pagesExperiment 9 Total Serum CholesterolGneiss Louie Gem AlmazanNo ratings yet
- Characterization of Saponifiable Lipids (Melted Fat, Lecithin, and Plant Oils) Using Grease-Spot, Saponification and Unsaturation TestsDocument17 pagesCharacterization of Saponifiable Lipids (Melted Fat, Lecithin, and Plant Oils) Using Grease-Spot, Saponification and Unsaturation TestsEzra TimothyNo ratings yet
- 2nd Sem Food Science NotesDocument46 pages2nd Sem Food Science Notesponavnit85% (13)
- EXPERIMENT 1 Food ChemistryDocument9 pagesEXPERIMENT 1 Food ChemistryNabila Husna100% (2)
- Emulsion: # What Is Emulsion? How Emulsion Prepared by Dry Gum Method and Wet Gum MethodDocument8 pagesEmulsion: # What Is Emulsion? How Emulsion Prepared by Dry Gum Method and Wet Gum MethodMahesh Phad100% (1)
- Efficient LiquifactionDocument10 pagesEfficient LiquifactionMuhammadTalalNooriNo ratings yet
- Pygas (Pyrolysis Gasoline)Document5 pagesPygas (Pyrolysis Gasoline)sanjeevvange100% (1)
- Chem Lab Final Exam Notes 2Document7 pagesChem Lab Final Exam Notes 2Jaira Emmarina100% (1)
- Formol TitrationDocument11 pagesFormol TitrationWinda EngkesaNo ratings yet
- Qualitative Analysis of CarbohydrateDocument13 pagesQualitative Analysis of CarbohydrateJulio Francisco100% (2)
- Qualitative Test of Lipids IIDocument18 pagesQualitative Test of Lipids IIManohar Pattar75% (4)
- LIPIDS D PharmDocument65 pagesLIPIDS D PharmMadhuri poulkarNo ratings yet
- FOOD CHEMISTRY CARBOHYDRATES BY DR. BOOMINATHAN - PPT I Lecture 1.august.2012Document49 pagesFOOD CHEMISTRY CARBOHYDRATES BY DR. BOOMINATHAN - PPT I Lecture 1.august.2012Abdiqani Mohamed Adan100% (1)
- Saponification Practical ReportDocument6 pagesSaponification Practical ReportGilbert NdibeNo ratings yet
- Qualitative Test of Lipids Unsaturation Test For LipidsDocument12 pagesQualitative Test of Lipids Unsaturation Test For LipidsQasmNo ratings yet
- Final Report Expt 2Document6 pagesFinal Report Expt 2Cha CanceranNo ratings yet
- Av of OilDocument3 pagesAv of OilOmotemisola AdegbiteNo ratings yet
- Experiment 16 Determination of Free Fatty Acids and Acid Value in Oils and FatsDocument2 pagesExperiment 16 Determination of Free Fatty Acids and Acid Value in Oils and FatspthimanshuNo ratings yet
- Department of Food Engineering Fe376 Food Quality Control FATS AND OILS (First Education, Group G5)Document8 pagesDepartment of Food Engineering Fe376 Food Quality Control FATS AND OILS (First Education, Group G5)SevilayNo ratings yet
- Analytical Chemistry Notes IiDocument9 pagesAnalytical Chemistry Notes IiJabez MatigaNo ratings yet
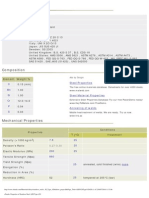
- Stainless Steel AISI Type 420Document2 pagesStainless Steel AISI Type 420Samir SalamaNo ratings yet
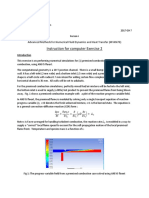
- AdvanceCFD ComputerExercise 2 2017Document5 pagesAdvanceCFD ComputerExercise 2 2017Akash RamannNo ratings yet
- Thermal PollutionDocument15 pagesThermal PollutionArunniya SNo ratings yet
- SsseeeDocument9 pagesSsseeeboom rangNo ratings yet
- Atul Limited: Polymers DivisionDocument2 pagesAtul Limited: Polymers Divisionsantosh mhetreNo ratings yet
- Duo Cone SealDocument15 pagesDuo Cone SealM Ferry AnwarNo ratings yet
- Plasma DrillingDocument31 pagesPlasma DrillingSaurav SenguptaNo ratings yet
- NCERT Solutions For Class 7 Science Chapter 6Document4 pagesNCERT Solutions For Class 7 Science Chapter 6raju bhowalNo ratings yet
- Vinyblan Brochure Jan 2018RSDocument10 pagesVinyblan Brochure Jan 2018RSAPEX SONNo ratings yet
- Definition of Modelling and SimulationDocument2 pagesDefinition of Modelling and Simulationjokish100% (1)
- Designing Analog ChipsDocument206 pagesDesigning Analog Chips33angleNo ratings yet
- Jan 2022 Chem Unit 3 MsDocument26 pagesJan 2022 Chem Unit 3 MsSyeda SadiaNo ratings yet
- Environmental PollutionDocument6 pagesEnvironmental PollutionLalit KapuraniNo ratings yet
- Environmental Pollution Control (ET ZC362 - WILP Course) : BITS PilaniDocument41 pagesEnvironmental Pollution Control (ET ZC362 - WILP Course) : BITS Pilanisa_arunkumarNo ratings yet
- Survey and Risk Assessment of Whitening ProductsDocument96 pagesSurvey and Risk Assessment of Whitening ProductsenNo ratings yet
- Din 17175 PDFDocument22 pagesDin 17175 PDFluap161256No ratings yet
- Liquid Solution-04 - Assignments (N)Document16 pagesLiquid Solution-04 - Assignments (N)Raju SinghNo ratings yet
- Translucency of Human EnamelDocument5 pagesTranslucency of Human EnamelsmritinarayanNo ratings yet
- Marine Chemistry: 10.1016/j.marchem.2013.02.006Document46 pagesMarine Chemistry: 10.1016/j.marchem.2013.02.006Usman AliNo ratings yet
- Iso 2719-2016Document11 pagesIso 2719-2016Jozsef FazekadNo ratings yet
- MSDS - Finish SA 650 1006Document4 pagesMSDS - Finish SA 650 1006salman KhanNo ratings yet
- BTech IT 2018-Onwards Jun-2021Document200 pagesBTech IT 2018-Onwards Jun-2021KittyPantherNo ratings yet
- Start A Phenyl Making Business in IndiaDocument11 pagesStart A Phenyl Making Business in IndiaasdfNo ratings yet
- Purified Water System Validation - 1Document9 pagesPurified Water System Validation - 1sarada jena100% (1)
- Chapter 4. The Chemistry of HydrogenDocument28 pagesChapter 4. The Chemistry of HydrogenyosefNo ratings yet
- ABR Radiologic Physics Initial Certification Study Guide: Computer-Based ExaminationsDocument4 pagesABR Radiologic Physics Initial Certification Study Guide: Computer-Based ExaminationsEd ShieldsNo ratings yet
- 44Document1 page44snehal medheNo ratings yet
- Phosphor Bronze CuSn6-PB103 Datasheet PDFDocument1 pagePhosphor Bronze CuSn6-PB103 Datasheet PDFRemo StortiniNo ratings yet
- International Chemistry Olympiad Problems Volume 01 (1968-1988)Document408 pagesInternational Chemistry Olympiad Problems Volume 01 (1968-1988)Science Olympiad Blog100% (5)
- Guidelines for Integrating Management Systems and Metrics to Improve Process Safety PerformanceFrom EverandGuidelines for Integrating Management Systems and Metrics to Improve Process Safety PerformanceNo ratings yet
- Functional Safety from Scratch: A Practical Guide to Process Industry ApplicationsFrom EverandFunctional Safety from Scratch: A Practical Guide to Process Industry ApplicationsNo ratings yet
- Sodium Bicarbonate: Nature's Unique First Aid RemedyFrom EverandSodium Bicarbonate: Nature's Unique First Aid RemedyRating: 5 out of 5 stars5/5 (21)
- Guidelines for the Management of Change for Process SafetyFrom EverandGuidelines for the Management of Change for Process SafetyNo ratings yet
- Bioinspired Materials Science and EngineeringFrom EverandBioinspired Materials Science and EngineeringGuang YangNo ratings yet
- Troubleshooting Process Plant Control: A Practical Guide to Avoiding and Correcting MistakesFrom EverandTroubleshooting Process Plant Control: A Practical Guide to Avoiding and Correcting MistakesRating: 1 out of 5 stars1/5 (2)
- Process Plant Equipment: Operation, Control, and ReliabilityFrom EverandProcess Plant Equipment: Operation, Control, and ReliabilityRating: 5 out of 5 stars5/5 (1)
- Guidelines for Chemical Process Quantitative Risk AnalysisFrom EverandGuidelines for Chemical Process Quantitative Risk AnalysisRating: 5 out of 5 stars5/5 (1)
- Polyoxymethylene Handbook: Structure, Properties, Applications and their NanocompositesFrom EverandPolyoxymethylene Handbook: Structure, Properties, Applications and their NanocompositesNo ratings yet
- Soap Manufacturing TechnologyFrom EverandSoap Manufacturing TechnologyLuis SpitzRating: 4 out of 5 stars4/5 (6)
- The Perfumed Pages of History: A Textbook on Fragrance CreationFrom EverandThe Perfumed Pages of History: A Textbook on Fragrance CreationRating: 4 out of 5 stars4/5 (1)
- A New Approach to HAZOP of Complex Chemical ProcessesFrom EverandA New Approach to HAZOP of Complex Chemical ProcessesNo ratings yet
- Life Cycle of a Process PlantFrom EverandLife Cycle of a Process PlantMahdi NouriNo ratings yet
- Bow Ties in Risk Management: A Concept Book for Process SafetyFrom EverandBow Ties in Risk Management: A Concept Book for Process SafetyNo ratings yet
- Chemical Process Equipment - Selection and Design (Revised 2nd Edition)From EverandChemical Process Equipment - Selection and Design (Revised 2nd Edition)Rating: 5 out of 5 stars5/5 (3)
- Well Control for Completions and InterventionsFrom EverandWell Control for Completions and InterventionsRating: 4 out of 5 stars4/5 (10)