You might also like
- SN and E 2019 PDFDocument108 pagesSN and E 2019 PDFDwi ShabrinaNo ratings yet
- Dhoom # 9 Haloalkane & Haloarene in One Shot (10.6.2020)Document156 pagesDhoom # 9 Haloalkane & Haloarene in One Shot (10.6.2020)Jeet RathodNo ratings yet
- 11 Chemistry Notes Chapter 13Document23 pages11 Chemistry Notes Chapter 13Deevanshi MalhotraNo ratings yet
- Chapter 4 SUBSTITUTION REACTIONDocument35 pagesChapter 4 SUBSTITUTION REACTIONHalimatun MustafaNo ratings yet
- 100S120 CS16 IoyDocument34 pages100S120 CS16 Ioyb101112154No ratings yet
- CH - 13Document23 pagesCH - 13Preet RedduNo ratings yet
- Chapter 10 PDFDocument10 pagesChapter 10 PDFKelsi Kyla PeraltaNo ratings yet
- Recall Alkyl Halides (Haloalkanes) : The Polarity and Strength of The C-X BondDocument32 pagesRecall Alkyl Halides (Haloalkanes) : The Polarity and Strength of The C-X BondafafNo ratings yet
- CHAPTER3Document29 pagesCHAPTER3Laura BeltranNo ratings yet
- Chapter 6 Nucleophilic Substitution On Saturated CarbonsDocument46 pagesChapter 6 Nucleophilic Substitution On Saturated CarbonsNUR HAZWANI BINTI MOHAMAD SANI / UPMNo ratings yet
- OCI Lecture4-5Document19 pagesOCI Lecture4-5Baga DagaNo ratings yet
- Aliphatic HydroCarbonDocument34 pagesAliphatic HydroCarbonSuparom ManijutakornNo ratings yet
- Chapter 13Document28 pagesChapter 13Kushagardrall DrallNo ratings yet
- Complete Organic Chemistry (Brahmastra) Part 2Document763 pagesComplete Organic Chemistry (Brahmastra) Part 2mohdamaankhan74No ratings yet
- AlkenesDocument30 pagesAlkenesapi-3734333No ratings yet
- 14.0 Haloalkanes - 02Document4 pages14.0 Haloalkanes - 02Mumtaz BarhiyaNo ratings yet
- Hydrolysis 2016Document20 pagesHydrolysis 2016Kashish ParmarNo ratings yet
- ၁၀တန်းOrganic chemistry summaryDocument6 pages၁၀တန်းOrganic chemistry summarySANLU HTUTNo ratings yet
- Reaction Mechanism - 15.3.2020 New PDFDocument20 pagesReaction Mechanism - 15.3.2020 New PDFsushant mohodNo ratings yet
- Haloalkanes & HaloarenesDocument10 pagesHaloalkanes & Haloarenesakshatshukla2021No ratings yet
- Alkyl Halide PDFDocument21 pagesAlkyl Halide PDFLitmus GodNo ratings yet
- Synthesis of AlkynesDocument31 pagesSynthesis of AlkynesttinbddinNo ratings yet
- Organic Chemistry: CollegeDocument34 pagesOrganic Chemistry: CollegeArwa AhmedNo ratings yet
- Alkenes 1Document33 pagesAlkenes 1Abdullah AmjadNo ratings yet
- Alkenes: CC H H H HDocument39 pagesAlkenes: CC H H H Hmanali shahNo ratings yet
- 8 Alkene & AlkyneDocument74 pages8 Alkene & Alkynerusnah chungNo ratings yet
- 20 Alkyl Halide Revision Notes QuizrrDocument78 pages20 Alkyl Halide Revision Notes QuizrrMONEY ALLNo ratings yet
- Chapter 3Document24 pagesChapter 3Eshita SharmaNo ratings yet
- Organic Chemistry IIDocument6 pagesOrganic Chemistry IIRoberto SIlvaNo ratings yet
- Alkenes 7: Oxidation of Alkenes: Chemistry 318/310M SesslerDocument18 pagesAlkenes 7: Oxidation of Alkenes: Chemistry 318/310M Sesslerwhidow1973No ratings yet
- Reaction of Organic Compound Chapter-2Document13 pagesReaction of Organic Compound Chapter-2Fikere'ab HabtamuNo ratings yet
- Organic ChemistryDocument7 pagesOrganic ChemistryABDULLAH SHAHZADNo ratings yet
- Substitution Elimination (Alkilhalides) Bab 5 FessendenDocument113 pagesSubstitution Elimination (Alkilhalides) Bab 5 Fessendenahmad jamalNo ratings yet
- Halogeno AlkanesDocument12 pagesHalogeno AlkanessaraNo ratings yet
- CBSE Class 12 Chem Notes Question Bank Haloalkanes and Haloarenes PDFDocument18 pagesCBSE Class 12 Chem Notes Question Bank Haloalkanes and Haloarenes PDFhehe11No ratings yet
- Halogen Derivatives: B.SC Part - 1 (Honour's and Subsidiary)Document18 pagesHalogen Derivatives: B.SC Part - 1 (Honour's and Subsidiary)Lokesh BorseNo ratings yet
- Ch11 Self-Study PDFDocument22 pagesCh11 Self-Study PDFRida Naila MangiNo ratings yet
- Anic Chemistry Alkyl HalidesDocument15 pagesAnic Chemistry Alkyl Halideseamcetmaterials100% (6)
- Halogeno CompoundsDocument28 pagesHalogeno Compoundsapi-3734333No ratings yet
- Hydrocarbons A-7Document4 pagesHydrocarbons A-7REJA MUKIB KHANNo ratings yet
- Chemistry - Sample Question Paper - 9Document6 pagesChemistry - Sample Question Paper - 9Mohd AdilNo ratings yet
- 5B2-5D Chapter5 bwk10303Document41 pages5B2-5D Chapter5 bwk10303Zulhazuwanah ZolekafeliNo ratings yet
- 2016 Lect6a Substitution Elimination (Alkilhalides)Document144 pages2016 Lect6a Substitution Elimination (Alkilhalides)Bryan SuryapranataNo ratings yet
- Hydrocarbon MAIN NotesDocument25 pagesHydrocarbon MAIN NotesHamad FarooqueNo ratings yet
- Haloalkanes Haloarnes NotesDocument44 pagesHaloalkanes Haloarnes Noteshareharanbt22No ratings yet
- Topic 10 Alkane TutorialDocument6 pagesTopic 10 Alkane TutorialTimNo ratings yet
- Hydrocarbon Notes - Till AlkaneDocument8 pagesHydrocarbon Notes - Till AlkaneHamad FarooqueNo ratings yet
- HALOALKANESDocument87 pagesHALOALKANES09. Bishesta Siwakoti 10 BNo ratings yet
- Chapter 3Document61 pagesChapter 3Avy VyNo ratings yet
- PDFDocument12 pagesPDFJhonsonNo ratings yet
- Steam Cracking Olefins PDFDocument47 pagesSteam Cracking Olefins PDFeltipovalencia100% (1)
- Hydrocarbons NotesDocument13 pagesHydrocarbons NotesShivansh Pundir100% (1)
- Organic Chemistry MinDocument18 pagesOrganic Chemistry Mina711789322No ratings yet
- (L8) - (JLD 2.0) - Reaction Mechanisms - 17 SeptDocument55 pages(L8) - (JLD 2.0) - Reaction Mechanisms - 17 Septchirag birlaNo ratings yet
- Matriculation Chemistry (Haloalkane)Document46 pagesMatriculation Chemistry (Haloalkane)ridwanNo ratings yet
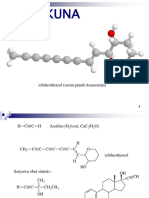
- Alkuna: Ichtheothereol (Racun Panah Amazonian)Document15 pagesAlkuna: Ichtheothereol (Racun Panah Amazonian)Anita puspitasariNo ratings yet
- Class 11 Chemistry Revision Notes HydrocarbonsDocument18 pagesClass 11 Chemistry Revision Notes HydrocarbonsSURESH SURESHNo ratings yet
- 03-Neet12-s2-Te C+P+B - Questions - m1Document32 pages03-Neet12-s2-Te C+P+B - Questions - m1VishruthNo ratings yet
- Preparations of Alkanes, Alkenes, AlkynesDocument71 pagesPreparations of Alkanes, Alkenes, AlkynesRavi100% (1)
- 5 StereoisomersDocument37 pages5 StereoisomersAudrey Patrick KallaNo ratings yet
- UN 2017 Kimia PDFDocument17 pagesUN 2017 Kimia PDFAnonymous vDMrfyYqjoNo ratings yet
- Konformasi Dan Stereokimia: Octane OctaneDocument137 pagesKonformasi Dan Stereokimia: Octane OctaneAnonymous vDMrfyYqjoNo ratings yet
- Un 2013 Kimia PDFDocument16 pagesUn 2013 Kimia PDFAnonymous vDMrfyYqjoNo ratings yet
- Chemical Reaction Stochiometrymissen SmithDocument48 pagesChemical Reaction Stochiometrymissen SmithTaufik AchmadNo ratings yet
- Kesetimbangan KimiaDocument44 pagesKesetimbangan KimiaAnonymous vDMrfyYqjoNo ratings yet
- Chemical Reaction Stochiometrymissen SmithDocument48 pagesChemical Reaction Stochiometrymissen SmithTaufik AchmadNo ratings yet
- Substitusi Aromatik Materi Kimia PDFDocument10 pagesSubstitusi Aromatik Materi Kimia PDFAnonymous vDMrfyYqjoNo ratings yet
- Konformasi Dan Stereokimia: Octane OctaneDocument137 pagesKonformasi Dan Stereokimia: Octane OctaneAnonymous vDMrfyYqjoNo ratings yet
- 5 StereoisomersDocument37 pages5 StereoisomersAudrey Patrick KallaNo ratings yet
- MacromoleculesDocument46 pagesMacromoleculesLady Ayessa AllonNo ratings yet
- Chapter 2 Pharmaceutical Aids and NecessitiesDocument11 pagesChapter 2 Pharmaceutical Aids and NecessitiesZarah Pauline Jimenez100% (2)
- Ethers R-O-R or R-O-R : NomenclatureDocument17 pagesEthers R-O-R or R-O-R : NomenclatureAbhishek Guddad100% (1)
- Subject: Biochemistry Assignment On: LipidsDocument8 pagesSubject: Biochemistry Assignment On: LipidsMehran AhmadNo ratings yet
- Cambridge IGCSE ™: Chemistry 0620/43Document11 pagesCambridge IGCSE ™: Chemistry 0620/43Shashwat VignaradjNo ratings yet
- (QUIZ) Chemistry of Carbohydrates (LEC)Document8 pages(QUIZ) Chemistry of Carbohydrates (LEC)Abdul JackowlNo ratings yet
- Calculating Atomic and Molecular MassesDocument30 pagesCalculating Atomic and Molecular MassesBright MindsNo ratings yet
- Naming CompoundsDocument9 pagesNaming CompoundsDecena VillanuevaNo ratings yet
- Salt Analysis (Mega)Document40 pagesSalt Analysis (Mega)Anant JainNo ratings yet
- Physical Science 4Document20 pagesPhysical Science 4Christine AtencioNo ratings yet
- Testbank3 Syror Baser+svar PDFDocument10 pagesTestbank3 Syror Baser+svar PDFSandileVilaneNo ratings yet
- Basic Concept of Organic Chemstry1 1Document40 pagesBasic Concept of Organic Chemstry1 1Raj ShresthaNo ratings yet
- Chapter 6, 7 Halohydrocarbon, Alcohol, PhenolDocument89 pagesChapter 6, 7 Halohydrocarbon, Alcohol, PhenolGan Suk Ling100% (1)
- Nomenclature of AlkenesDocument3 pagesNomenclature of AlkenesMohamad AzaniNo ratings yet
- Tests For Categorizing Carboxylic Acids and Their DerivativesDocument7 pagesTests For Categorizing Carboxylic Acids and Their DerivativesFaye ConstantinoNo ratings yet
- Chemistry SPMDocument25 pagesChemistry SPMSudhan NairNo ratings yet
- Class 10 Science PaperDocument1 pageClass 10 Science PapervirendraNo ratings yet
- Group Vii ElementsDocument22 pagesGroup Vii ElementsJohn KibuukaNo ratings yet
- IGCSE Biology Chapter 4: Biological MoleculesDocument7 pagesIGCSE Biology Chapter 4: Biological MoleculesNabil Ahmed0% (1)
- Engineering Chemistry Lab ManualDocument25 pagesEngineering Chemistry Lab ManualmsdineshpaiNo ratings yet
- Haloalkanes and Haloarenes by Bharat Panchal CBSE 2022 Term 1Document25 pagesHaloalkanes and Haloarenes by Bharat Panchal CBSE 2022 Term 1roceni100% (2)
- 9701 s15 QP 11 PDFDocument16 pages9701 s15 QP 11 PDFAl BeruniNo ratings yet
- Cambridge IGCSE™ Chemistry 4th Edition (Bryan Earl, Doug Wilford) (Z-Library)Document18 pagesCambridge IGCSE™ Chemistry 4th Edition (Bryan Earl, Doug Wilford) (Z-Library)Muhammad TayyabNo ratings yet
- Unit 9 PDFDocument16 pagesUnit 9 PDFcarlette11No ratings yet
- Acids and Bases Review MCQDocument2 pagesAcids and Bases Review MCQAnna DixonNo ratings yet
- CHEM 151 (Quiz 1 (Make-Up) Key)Document3 pagesCHEM 151 (Quiz 1 (Make-Up) Key)Chantel AceveroNo ratings yet
- Chemistry Lab Practical For Students of Class XII PDFDocument8 pagesChemistry Lab Practical For Students of Class XII PDFHendrickNo ratings yet
- Basic Terminology: do-24CN +2Document4 pagesBasic Terminology: do-24CN +2vinayaksharma1911No ratings yet
- AS RedoxDocument20 pagesAS RedoxIbrahim AbidNo ratings yet