You might also like
- Annual Reports in Organic Synthesis — 1971From EverandAnnual Reports in Organic Synthesis — 1971John McMurryNo ratings yet
- 20 Alkyl Halide Revision Notes QuizrrDocument78 pages20 Alkyl Halide Revision Notes QuizrrMONEY ALLNo ratings yet
- Presentation Alkyl HalidesDocument20 pagesPresentation Alkyl HalidesCaroline MuthoniNo ratings yet
- 4 June 2022 ChemistryDocument68 pages4 June 2022 Chemistryadsaks2528No ratings yet
- Alkyl HalidesDocument26 pagesAlkyl Halidesharerambaghel906No ratings yet
- Haloalkanes and Haloarenes1Document15 pagesHaloalkanes and Haloarenes1Poorni RenuNo ratings yet
- Chapter 6-Alkylhalide 145 PDFDocument33 pagesChapter 6-Alkylhalide 145 PDFKishore KishoreNo ratings yet
- Topic 6 - Alkyl Halide and Carbonyl Compounds Organic Compounds Containing A HalogenDocument11 pagesTopic 6 - Alkyl Halide and Carbonyl Compounds Organic Compounds Containing A HalogenamitNo ratings yet
- Dhoom # 9 Haloalkane & Haloarene in One Shot (10.6.2020)Document156 pagesDhoom # 9 Haloalkane & Haloarene in One Shot (10.6.2020)Jeet RathodNo ratings yet
- Hydrocarbons: Alkanes and Their PropertiesDocument18 pagesHydrocarbons: Alkanes and Their PropertiesSURESH SURESHNo ratings yet
- 4alkyl Halides and AlcoholsDocument85 pages4alkyl Halides and AlcoholssharmimiameerasanadyNo ratings yet
- Organohalogens ExplainedDocument13 pagesOrganohalogens ExplainedcikguhafidzuddinNo ratings yet
- Haloalkanes Haloarnes NotesDocument44 pagesHaloalkanes Haloarnes Noteshareharanbt22No ratings yet
- 12 Chemistry Haloalkanes and Haloarenes Test 05 Answer s2l6 PDFDocument2 pages12 Chemistry Haloalkanes and Haloarenes Test 05 Answer s2l6 PDFShreyash KolekarNo ratings yet
- 11 Chemistry Notes Chapter 13Document23 pages11 Chemistry Notes Chapter 13Deevanshi MalhotraNo ratings yet
- Chapter 13Document28 pagesChapter 13Kushagardrall DrallNo ratings yet
- Organic Name ReactionsDocument2 pagesOrganic Name ReactionsPratham ZalaNo ratings yet
- ABC 3 (Theory Exercise)Document11 pagesABC 3 (Theory Exercise)Mayank GoyalNo ratings yet
- Alkyl Halide and Aryl HalideDocument43 pagesAlkyl Halide and Aryl HalideShivanshi0950% (2)
- Halogeno AlkanesDocument12 pagesHalogeno AlkanessaraNo ratings yet
- Lecture9 Alkenes2010Document79 pagesLecture9 Alkenes2010Inoxcent MoonNo ratings yet
- Anic Chemistry Alkyl HalidesDocument15 pagesAnic Chemistry Alkyl Halideseamcetmaterials100% (6)
- CH - 13Document23 pagesCH - 13Preet RedduNo ratings yet
- Alkyne-Anna IN CLASS 1Document26 pagesAlkyne-Anna IN CLASS 1Siti Farhanah Mohd NasirNo ratings yet
- Xicbse Che Asst 1 Ans (1)Document3 pagesXicbse Che Asst 1 Ans (1)tanishkakannan3253No ratings yet
- Chapter 4 SUBSTITUTION REACTIONDocument35 pagesChapter 4 SUBSTITUTION REACTIONHalimatun MustafaNo ratings yet
- Haloalkanes and Haloarenes: Day ThirtyDocument16 pagesHaloalkanes and Haloarenes: Day Thirtyfastestnews12No ratings yet
- Organic Chemistry MinDocument18 pagesOrganic Chemistry Mina711789322No ratings yet
- Electrophilic Aromatic SubstitutionDocument5 pagesElectrophilic Aromatic Substitutioneman mamdohNo ratings yet
- Haloalkanes and HaloarenesDocument14 pagesHaloalkanes and Haloarenesshreyansh tanwarNo ratings yet
- 20 Alkyl Halide Revision Notes Getmarks AppDocument79 pages20 Alkyl Halide Revision Notes Getmarks AppArnav GuptaNo ratings yet
- Types and Reactions of Alkyl HalidesDocument68 pagesTypes and Reactions of Alkyl HalidesAniruddha KawadeNo ratings yet
- Alkyl and Aryl Halide ClassificationDocument21 pagesAlkyl and Aryl Halide ClassificationLitmus GodNo ratings yet
- Alkyl HalidesDocument15 pagesAlkyl HalidesDjdj DjdjNo ratings yet
- 02 Hydrogen Jeemain - GuruDocument21 pages02 Hydrogen Jeemain - GuruSarala KakavakamNo ratings yet
- Haloalkanes and HaloarenesDocument14 pagesHaloalkanes and HaloarenesKalpesh BishnoiNo ratings yet
- 11 Hydrocarbon Study NotesDocument23 pages11 Hydrocarbon Study NotesVivek KumarNo ratings yet
- Edit 1592206021 195648a81951e931 Alkyl Halide - TheoryDocument23 pagesEdit 1592206021 195648a81951e931 Alkyl Halide - Theorysultaneaditya.1No ratings yet
- Organic Name ReactionsDocument2 pagesOrganic Name ReactionsShruti MohrilNo ratings yet
- Alkyl Halides: Organic ChemistryDocument35 pagesAlkyl Halides: Organic ChemistryBlessy MartinNo ratings yet
- Hydrocarbon LatestDocument23 pagesHydrocarbon LatestHimanshuNo ratings yet
- Organic TuteDocument1 pageOrganic TuteDimuthu SandaruwanNo ratings yet
- Haloalkanes & HaloarenesDocument10 pagesHaloalkanes & Haloarenesakshatshukla2021No ratings yet
- CH CH CH CH I: BRCH CH CH CCH BR CH CHDocument24 pagesCH CH CH CH I: BRCH CH CH CCH BR CH CHSam TabujaraNo ratings yet
- Chemistry: Chapter - 10 Haloalkanes and HaloarenesDocument10 pagesChemistry: Chapter - 10 Haloalkanes and HaloarenesAjith 007No ratings yet
- Organic - Chemistry HandoutsDocument34 pagesOrganic - Chemistry HandoutsVernice OrtegaNo ratings yet
- Hydrocarbons: AlkanesDocument12 pagesHydrocarbons: AlkanesFredrick HeffersonNo ratings yet
- SN and E 2019 PDFDocument108 pagesSN and E 2019 PDFDwi ShabrinaNo ratings yet
- P-101 AlkynesDocument10 pagesP-101 AlkynesNISARG PATKARNo ratings yet
- Chapter 5 Alkyl HalidesDocument33 pagesChapter 5 Alkyl HalidesKonoli NuingNo ratings yet
- 14.0 Haloalkanes - 02Document4 pages14.0 Haloalkanes - 02Mumtaz BarhiyaNo ratings yet
- HydrocarbonsDocument37 pagesHydrocarbonsraghavsuresh865No ratings yet
- Alkyl Halides & Aryl Halides: Victor GrignardDocument50 pagesAlkyl Halides & Aryl Halides: Victor GrignardsarahNo ratings yet
- Carboxylic Acid & Acid Derivatives and Amines: Exercise - IDocument3 pagesCarboxylic Acid & Acid Derivatives and Amines: Exercise - ILifestyle BoomNo ratings yet
- Chapter 2-2Document29 pagesChapter 2-2AdellNo ratings yet
- Problems 11-20-06 Chem231-SSCC: C H H C C C H CH C H CL C C H CH C H + CLDocument6 pagesProblems 11-20-06 Chem231-SSCC: C H H C C C H CH C H CL C C H CH C H + CLsarahNo ratings yet
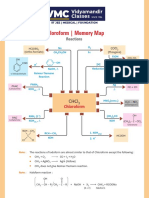
- Chloroform - Memory Map: ReactionsDocument1 pageChloroform - Memory Map: ReactionsAryan GuptaNo ratings yet
- Halogenation of Alkanes: Regioselectivity and MechanismDocument98 pagesHalogenation of Alkanes: Regioselectivity and MechanismAnonymous vDMrfyYqjoNo ratings yet
- Class - 12/chemistry-2 - Reactions of AlcoholsDocument60 pagesClass - 12/chemistry-2 - Reactions of AlcoholsAISHA AHAMMEDNo ratings yet
- Medical Device LogbookDocument86 pagesMedical Device LogbookafafNo ratings yet
- PIDAC Cleaning Disinfection and Sterilization 2013Document117 pagesPIDAC Cleaning Disinfection and Sterilization 2013georgeNo ratings yet
- Properties of Enzyme Inhibition (CH 3, 7)Document18 pagesProperties of Enzyme Inhibition (CH 3, 7)afaf100% (1)
- Electrical Safety ManualDocument63 pagesElectrical Safety Manualsuryavenkat_79No ratings yet
- Nucleic Acids (Patrick CH 6) : RNA & DNA - Structure & Intro To FunctionDocument16 pagesNucleic Acids (Patrick CH 6) : RNA & DNA - Structure & Intro To FunctionafafNo ratings yet
- Hydrophobicity: Physicochemical Properties of Drugs (CH 18)Document35 pagesHydrophobicity: Physicochemical Properties of Drugs (CH 18)afafNo ratings yet
- Drug Discovery from Natural SourcesDocument51 pagesDrug Discovery from Natural SourcesafafNo ratings yet
- 5ii PDFDocument13 pages5ii PDFafafNo ratings yet
- 1 IIDocument9 pages1 IIafafNo ratings yet
- 3 II PDFDocument5 pages3 II PDFafafNo ratings yet
- Wk6 CBMS103backgroundDocument40 pagesWk6 CBMS103backgroundafafNo ratings yet
- 7 II PDFDocument42 pages7 II PDFafafNo ratings yet
- 9 II PDFDocument8 pages9 II PDFafafNo ratings yet
- 8 II PDFDocument9 pages8 II PDFafafNo ratings yet
- W 1Document2 pagesW 1afafNo ratings yet
- 7Document12 pages7afafNo ratings yet
- Recall Alkyl Halides (Haloalkanes) : The Polarity and Strength of The C-X BondDocument32 pagesRecall Alkyl Halides (Haloalkanes) : The Polarity and Strength of The C-X BondafafNo ratings yet
- The Origin of The "Quartet" in Problem 29 (Dibenzyl Sulfoxide) - Set Two of Spectral Problems - ACT Workshop Two, in The Final Week of SemesterDocument4 pagesThe Origin of The "Quartet" in Problem 29 (Dibenzyl Sulfoxide) - Set Two of Spectral Problems - ACT Workshop Two, in The Final Week of SemesterafafNo ratings yet
- W 1Document2 pagesW 1afafNo ratings yet
- 7Document12 pages7afafNo ratings yet
- Recall Alkyl Halides (Haloalkanes) : The Polarity and Strength of The C-X BondDocument32 pagesRecall Alkyl Halides (Haloalkanes) : The Polarity and Strength of The C-X BondafafNo ratings yet
- Wk6 CBMS103backgroundDocument40 pagesWk6 CBMS103backgroundafafNo ratings yet
- 5&6 PDFDocument18 pages5&6 PDFafafNo ratings yet
- Lec 4 PDFDocument6 pagesLec 4 PDFafafNo ratings yet
- Lec 3Document8 pagesLec 3afafNo ratings yet
- Benzene and Aromaticity: John E. McmurryDocument40 pagesBenzene and Aromaticity: John E. McmurryafafNo ratings yet