You might also like
- Intro Thermo Public in Felt A PartialDocument67 pagesIntro Thermo Public in Felt A PartialWade WattsNo ratings yet
- Thermodynamic Models for Chemical Engineering: Design, Develop, Analyse and OptimizeFrom EverandThermodynamic Models for Chemical Engineering: Design, Develop, Analyse and OptimizeNo ratings yet
- Computational Methods for Process SimulationFrom EverandComputational Methods for Process SimulationRating: 3 out of 5 stars3/5 (1)
- MatPRO HelpDocument61 pagesMatPRO HelpnikanikolaeeNo ratings yet
- Vibrations of Power Plant Machines: A Guide for Recognition of Problems and TroubleshootingFrom EverandVibrations of Power Plant Machines: A Guide for Recognition of Problems and TroubleshootingNo ratings yet
- Chapter1 Thermodynamics PDFDocument24 pagesChapter1 Thermodynamics PDFsukandeNo ratings yet
- Physical Chemistry ModuleDocument122 pagesPhysical Chemistry ModuleFelix TinasheNo ratings yet
- Advanced Temperature Measurement and Control, Second EditionFrom EverandAdvanced Temperature Measurement and Control, Second EditionNo ratings yet
- The Chemical Reactor From Laboratory To Industrial Plant: Elio Santacesaria Riccardo TesserDocument574 pagesThe Chemical Reactor From Laboratory To Industrial Plant: Elio Santacesaria Riccardo TesserKoura KacouNo ratings yet
- 2015.462984.mechanical Engineering TextDocument545 pages2015.462984.mechanical Engineering Textcaserisimo hechoconamorNo ratings yet
- CRE Notes AMUDocument136 pagesCRE Notes AMUsatya.usct.900450No ratings yet
- Introduction To Controls: Chapter 1-5Document64 pagesIntroduction To Controls: Chapter 1-5BillChayangkulTanNo ratings yet
- Classical and Statisitcal ThermodinamicsDocument111 pagesClassical and Statisitcal ThermodinamicsitotoNo ratings yet
- Theory F 2012Document121 pagesTheory F 2012Ayesha FarooqNo ratings yet
- THSTMDocument85 pagesTHSTMPatrick SibandaNo ratings yet
- Genmix: A General Computer Program for Two-Dimensional Parabolic PhenomenaFrom EverandGenmix: A General Computer Program for Two-Dimensional Parabolic PhenomenaNo ratings yet
- Jj207 Thermodynamics 1 Grandcont 16 SeptDocument9 pagesJj207 Thermodynamics 1 Grandcont 16 SeptRaz MieNo ratings yet
- Nit and Dimensions Need To Include in This Document Able of OntentsDocument20 pagesNit and Dimensions Need To Include in This Document Able of OntentsSenthilkumaar JSNo ratings yet
- Chemical Thermodynamics (Ernő Keszei)Document362 pagesChemical Thermodynamics (Ernő Keszei)Ivan Sequera Grappin75% (4)
- Full Text 01Document72 pagesFull Text 01ogi sutrisnaNo ratings yet
- Introduction To Finite Element Method-University of Colorado at BoulderDocument513 pagesIntroduction To Finite Element Method-University of Colorado at Boulderfankenstain100% (2)
- Unit 1Document11 pagesUnit 1VINEET VYASNo ratings yet
- ME 311 Thermodynamics Course SyllabusDocument4 pagesME 311 Thermodynamics Course SyllabusSuga MilkNo ratings yet
- Chemical Kinetic Data Base For Combustion Chemistry. Part I. Methane and ReDocument192 pagesChemical Kinetic Data Base For Combustion Chemistry. Part I. Methane and ReMuhammad AtharNo ratings yet
- Embedded Mechatronic Systems 2: Analysis of Failures, Modeling, Simulation and OptimizationFrom EverandEmbedded Mechatronic Systems 2: Analysis of Failures, Modeling, Simulation and OptimizationNo ratings yet
- Thermo SyllabusDocument2 pagesThermo SyllabusDhenil ManubatNo ratings yet
- Engineering Thermodynamics Key ConceptsDocument57 pagesEngineering Thermodynamics Key ConceptsBasu SbNo ratings yet
- Chap 1Document16 pagesChap 1Hatem AbdelrahmanNo ratings yet
- Fundamental Concepts of Real GasdynamicsDocument37 pagesFundamental Concepts of Real GasdynamicsPatricio PedreiraNo ratings yet
- Engineering Thermodynamics Lecture Notes (A Very Rough Draft)Document34 pagesEngineering Thermodynamics Lecture Notes (A Very Rough Draft)kannanjbr100% (1)
- Physics 3 ReportDocument46 pagesPhysics 3 ReportNafih ZebariNo ratings yet
- Thermodynamics in Materials Science: Three Levels of the Thermodynamic ApparatusDocument17 pagesThermodynamics in Materials Science: Three Levels of the Thermodynamic ApparatusEre ValdesNo ratings yet
- Jha Dissertation 2021Document103 pagesJha Dissertation 2021Prateek VaishNo ratings yet
- Gas Extraction An Introduction To Fundamentals of Supercritical Fluids and The Application To Separation Processes by Prof. Dr.-Ing. Gerd Brunner (Auth.) PDFDocument396 pagesGas Extraction An Introduction To Fundamentals of Supercritical Fluids and The Application To Separation Processes by Prof. Dr.-Ing. Gerd Brunner (Auth.) PDFMUHAMMAD KHOLIDNo ratings yet
- Mathermatica Computer Programs For Physical Chemistry PDFDocument255 pagesMathermatica Computer Programs For Physical Chemistry PDFAmar Alansi100% (1)
- BITS F111 Thermodynamics Handout 2014-15Document2 pagesBITS F111 Thermodynamics Handout 2014-15shivaraj1996No ratings yet
- CHEMICAL PROCESS PLANTDocument92 pagesCHEMICAL PROCESS PLANTDhruv AgarwalNo ratings yet
- Automatic Controls for Heating and Air Conditioning: Principles and ApplicationsFrom EverandAutomatic Controls for Heating and Air Conditioning: Principles and ApplicationsRating: 4 out of 5 stars4/5 (2)
- Thermodynamics and The Simulation EngineerDocument43 pagesThermodynamics and The Simulation EngineerNeagu MihaelaNo ratings yet
- BestDocument15 pagesBestTaimoor100% (2)
- Engineering Thermodynamics Lecture Notes (Draft) : Wayne HackerDocument24 pagesEngineering Thermodynamics Lecture Notes (Draft) : Wayne Hackerrkadiraj7011No ratings yet
- Classical and Geometrical Theory of Chemical and Phase ThermodynamicsFrom EverandClassical and Geometrical Theory of Chemical and Phase ThermodynamicsNo ratings yet
- Physical Chemistry Laboratory ManualDocument123 pagesPhysical Chemistry Laboratory Manualveluselvamani100% (1)
- Applications of Quantum and Classical Connections in Modeling Atomic, Molecular and Electrodynamic SystemsFrom EverandApplications of Quantum and Classical Connections in Modeling Atomic, Molecular and Electrodynamic SystemsNo ratings yet
- Model Textbook of Chemistry For Senior Secodary Schools: January 2006Document499 pagesModel Textbook of Chemistry For Senior Secodary Schools: January 2006I am SheldonNo ratings yet
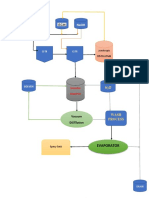
- Esquema Reactor de Metanol LaboratorioDocument33 pagesEsquema Reactor de Metanol LaboratoriochulinjavNo ratings yet
- Single-And Double-Bed Pressure Swing Adsorption Processes For H /CO Syngas SeparationDocument14 pagesSingle-And Double-Bed Pressure Swing Adsorption Processes For H /CO Syngas Separationkishna009No ratings yet
- BlockeDocument1 pageBlockekishna009No ratings yet
- Solving Method in HysysDocument2 pagesSolving Method in HysysChemsys Mail100% (2)
- Ansys LearningDocument22 pagesAnsys Learningkishna009No ratings yet
- Feasibility Analysis and Simulation of Argon Recovery in Low Oxygen-Purity Cryogenic Air Separation Process With Low Energy ConsumptionDocument25 pagesFeasibility Analysis and Simulation of Argon Recovery in Low Oxygen-Purity Cryogenic Air Separation Process With Low Energy Consumptionkishna009No ratings yet
- Fired Heater Optimization ISA ADDocument12 pagesFired Heater Optimization ISA ADNagaphani Kumar RavuriNo ratings yet
- Energy: Mehdi Mehrpooya, Mohammad Mehdi Moftakhari Sharifzadeh, Marc A. RosenDocument23 pagesEnergy: Mehdi Mehrpooya, Mohammad Mehdi Moftakhari Sharifzadeh, Marc A. Rosenkishna009No ratings yet
- Cryogenic Air SeparationDocument8 pagesCryogenic Air Separationkishna009No ratings yet
- Moderate Pressure ProcessDocument4 pagesModerate Pressure Processkishna009No ratings yet
- Design Sour Water Stripping Systems for Optimal Refinery OperationsDocument23 pagesDesign Sour Water Stripping Systems for Optimal Refinery Operationskishna009100% (2)
- Sizing Shell and Tube Heat ExchangerDocument17 pagesSizing Shell and Tube Heat ExchangerCallum Biggs100% (3)
- Vinasse Concentration and Juice Evaporation System IntegratedDocument39 pagesVinasse Concentration and Juice Evaporation System Integratedkishna009No ratings yet
- Energy Optimization in Parallel or Cross Feed Multiple-Effect EvaporatorDocument12 pagesEnergy Optimization in Parallel or Cross Feed Multiple-Effect Evaporatorkishna009No ratings yet
- Solving Method in HysysDocument2 pagesSolving Method in HysysChemsys Mail100% (2)
- (Wodek Gawronski) Advanced Structural Dynamics and (BookFi)Document436 pages(Wodek Gawronski) Advanced Structural Dynamics and (BookFi)Igor Di VaranoNo ratings yet
- Aspen Tech Compressor Modeling in Aspen PDFDocument16 pagesAspen Tech Compressor Modeling in Aspen PDFkishna009No ratings yet
- Energy Reduction Schemes For Multiple Effect Evaporator SystemsDocument10 pagesEnergy Reduction Schemes For Multiple Effect Evaporator Systemskishna009No ratings yet
- Distillation ch13Document14 pagesDistillation ch13kishna009No ratings yet
- Thermal Efficiency Fired HeaterDocument5 pagesThermal Efficiency Fired Heatermuhammad_asim_10No ratings yet
- SMR Life Cycle AssessmentDocument14 pagesSMR Life Cycle Assessmentkishna009100% (1)
- Reboiler 1Document12 pagesReboiler 1kishna009No ratings yet
- Fired Heater Optimization ISA ADDocument12 pagesFired Heater Optimization ISA ADNagaphani Kumar RavuriNo ratings yet
- Maria Syngas2MeOHDocument10 pagesMaria Syngas2MeOHRashveenaNo ratings yet
- Fired HeaterDocument28 pagesFired HeaterRapee PuaksungnoenNo ratings yet
- Heat Transfer in LTV FF EvaporatorDocument10 pagesHeat Transfer in LTV FF Evaporatorkishna009No ratings yet
- Single-And Double-Bed Pressure Swing Adsorption Processes For H /CO Syngas SeparationDocument14 pagesSingle-And Double-Bed Pressure Swing Adsorption Processes For H /CO Syngas Separationkishna009No ratings yet
- Effect of Cooling Water Temperature On MEEDocument8 pagesEffect of Cooling Water Temperature On MEEkishna009No ratings yet
- Falling Film Evaporator Application, Adv and Dis AdvDocument8 pagesFalling Film Evaporator Application, Adv and Dis Advkishna009No ratings yet
- Fuzzy Controller For MEEDocument6 pagesFuzzy Controller For MEEkishna009No ratings yet
- Loop Quantum GravityDocument69 pagesLoop Quantum GravityAnderson BernardiNo ratings yet
- Internship Report FACTDocument16 pagesInternship Report FACTBennetHailinkNo ratings yet
- Unit Ii, Lesson 4: Qualitative Research in Different Areas of KnowledgeDocument6 pagesUnit Ii, Lesson 4: Qualitative Research in Different Areas of KnowledgeJessy RoseNo ratings yet
- TextDocument17 pagesTextnaser zoabiNo ratings yet
- Replacing ATA5567/T5557/ TK5551 With ATA5577 Application NoteDocument5 pagesReplacing ATA5567/T5557/ TK5551 With ATA5577 Application NoteM0n3No ratings yet
- NetWorking Flashcards - QuizletDocument262 pagesNetWorking Flashcards - QuizletGB ReddyNo ratings yet
- ATTR State of The ArtDocument20 pagesATTR State of The ArtGenoveffa Ridalisi100% (1)
- Reservoir SedimentationDocument20 pagesReservoir SedimentationKathlienNo ratings yet
- SHS 2023 Thinking Skills Practice Test QuestionsDocument32 pagesSHS 2023 Thinking Skills Practice Test Questionsvenkatesh113No ratings yet
- Herman Pardamean Hutabarat: Mechanical EngineerDocument3 pagesHerman Pardamean Hutabarat: Mechanical EngineerHerman HutabaratNo ratings yet
- PH103 Section U Recap NotesDocument54 pagesPH103 Section U Recap NotesLuka MegurineNo ratings yet
- Grandparenting - Play With Me! Activities That Make Learning Fun 24 To 36 MonthsDocument3 pagesGrandparenting - Play With Me! Activities That Make Learning Fun 24 To 36 MonthsAmna ArshadNo ratings yet
- Sri Gopala Bhatta GoswamiDocument8 pagesSri Gopala Bhatta GoswamispshankarjpsNo ratings yet
- Introduction to measurement uncertainty components and calculationsDocument33 pagesIntroduction to measurement uncertainty components and calculationsLOUKILkarimNo ratings yet
- Mci 2005Document3 pagesMci 2005Christos LeptokaridisNo ratings yet
- Island of The Blue Dolphins: Before You Read The ChaptersDocument12 pagesIsland of The Blue Dolphins: Before You Read The ChaptersCostea CibanuNo ratings yet
- Works) : SABS 1200Document10 pagesWorks) : SABS 1200Palesa TshetlanyaneNo ratings yet
- Experiment 7 & 11: Presented By: Group 4Document55 pagesExperiment 7 & 11: Presented By: Group 4Julliane JuanNo ratings yet
- Microflow Air Sampler User ManualDocument31 pagesMicroflow Air Sampler User Manualira rahmaNo ratings yet
- LA36300 Circuit Breaker Data SheetDocument2 pagesLA36300 Circuit Breaker Data SheetALEJANDRO DOMINGUEZNo ratings yet
- New Correlations For Predicting Two Phase Electrical SubmersibleDocument11 pagesNew Correlations For Predicting Two Phase Electrical SubmersibleNicolás Ruiz FuentesNo ratings yet
- 2020 - (Sankaran) - Data Analytics in Reservoir EngineeringDocument107 pages2020 - (Sankaran) - Data Analytics in Reservoir EngineeringPedro100% (1)
- National Parks in IndiaDocument4 pagesNational Parks in IndiaVicky PandeyNo ratings yet
- Adobe Scan 11-Oct-2022Document5 pagesAdobe Scan 11-Oct-2022onkarkumarsolankiNo ratings yet
- Air Transportation ReviewerDocument16 pagesAir Transportation ReviewerJuly BacarNo ratings yet
- Gmat Clinic Course Notes PDFDocument213 pagesGmat Clinic Course Notes PDFDavid ThompsonNo ratings yet
- Family Nomenclature and Same-Name Divinities in Roman Religion and MythologyDocument17 pagesFamily Nomenclature and Same-Name Divinities in Roman Religion and MythologyhNo ratings yet
- Goody Anthropology of Sensess and Sensations LRF 2002 45 PDFDocument13 pagesGoody Anthropology of Sensess and Sensations LRF 2002 45 PDFAntroVirtualNo ratings yet
- Savage Worlds of Shadowrun FinalDocument29 pagesSavage Worlds of Shadowrun Finaljasonstierle100% (4)
- Electric Tool Parts List: Air Compressor 2007 - 04 - 02 Model EC 119 (E3)Document3 pagesElectric Tool Parts List: Air Compressor 2007 - 04 - 02 Model EC 119 (E3)Ivan GallegosNo ratings yet