You might also like
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (589)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (401)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (842)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (897)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5806)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1091)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- General Chemistry 2Document597 pagesGeneral Chemistry 2Orley G Fadriquel75% (4)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Pipe Alcorcon1Document86 pagesPipe Alcorcon1Nelson Naval Cabingas100% (11)
- Welding Practice For Duplex-1 PDFDocument18 pagesWelding Practice For Duplex-1 PDFBipin Rohit100% (1)
- Fluid Mechanics QtnsDocument3 pagesFluid Mechanics QtnsAnonymous NV1AeWXEo100% (1)
- Design of Rigid L Shaped Retaining Walls PDFDocument4 pagesDesign of Rigid L Shaped Retaining Walls PDFRajesh KumarNo ratings yet
- Design of Truss BridgeDocument18 pagesDesign of Truss BridgeBCE 2074 B Teacher100% (3)
- Structural Controls For Climate Responsive Design (For Cooling Purpose)Document31 pagesStructural Controls For Climate Responsive Design (For Cooling Purpose)mehtabhumikaaNo ratings yet
- Silicon Wafer FabricationDocument24 pagesSilicon Wafer FabricationJomel U. MaromaNo ratings yet
- Formal Report On Partial Molar Volume ExperimentDocument9 pagesFormal Report On Partial Molar Volume ExperimentBrandon Mutongorewa100% (2)
- Nehru Science Centre Services ReportDocument5 pagesNehru Science Centre Services ReportPranay Vira50% (2)
- MELCS For Bulletin in Science 10 Per QuarterDocument4 pagesMELCS For Bulletin in Science 10 Per QuarterMARIANNE SORIANO100% (1)
- The Extractive Industries and Society: Janpeter Schilling, Almut Schilling-Vacaflor, Riccarda Flemmer, Rebecca FroeseDocument12 pagesThe Extractive Industries and Society: Janpeter Schilling, Almut Schilling-Vacaflor, Riccarda Flemmer, Rebecca FroeseDicky Syaiful FadillahNo ratings yet
- Tabel Pull Up PDFDocument2 pagesTabel Pull Up PDFDicky Syaiful FadillahNo ratings yet
- Animal The International Journal of Animal BiosciencesDocument11 pagesAnimal The International Journal of Animal BiosciencesDicky Syaiful FadillahNo ratings yet
- Tabel Pull Up PDFDocument2 pagesTabel Pull Up PDFDicky Syaiful FadillahNo ratings yet
- Journal InternasionalDocument8 pagesJournal InternasionalDicky Syaiful FadillahNo ratings yet
- Computers & Education: Ling-Hsiu ChenDocument11 pagesComputers & Education: Ling-Hsiu ChenDicky Syaiful FadillahNo ratings yet
- Article in Press: Online Quiz Methods For Remedial Learning in Chemical EngineeringDocument7 pagesArticle in Press: Online Quiz Methods For Remedial Learning in Chemical EngineeringDicky Syaiful FadillahNo ratings yet
- CVX4343 Course MaterialDocument123 pagesCVX4343 Course MaterialnayaniNo ratings yet
- GPH 231Document113 pagesGPH 231Adwaitesh Aaradhya100% (2)
- 10 Penetration RateDocument30 pages10 Penetration RateMuhammad SaadNo ratings yet
- Safety Hazards: Section IVDocument17 pagesSafety Hazards: Section IVSooraj RajanNo ratings yet
- 52 - Model FP 43q BFDocument2 pages52 - Model FP 43q BFAdriano Moyses OutlookNo ratings yet
- Double-Effect Distillation and Thermal Integration Applied To The Ethanol Production ProcessDocument13 pagesDouble-Effect Distillation and Thermal Integration Applied To The Ethanol Production ProcessAhmad AsyraafNo ratings yet
- Unit-I. Soil ExplorationDocument139 pagesUnit-I. Soil ExplorationRwagatare civilcontractorsNo ratings yet
- Micro Analysis Report PDFDocument1 pageMicro Analysis Report PDFChalil FachroniNo ratings yet
- 258 - VeriVide Product Guide June09Document16 pages258 - VeriVide Product Guide June09Prabath De Silva0% (1)
- THERMODYNAMICS 1 - SEPT 2017 PRESENTATION Rev 1 PDFDocument28 pagesTHERMODYNAMICS 1 - SEPT 2017 PRESENTATION Rev 1 PDFVictor John PingkianNo ratings yet
- Essential Components of GC 1692116021Document53 pagesEssential Components of GC 1692116021Swapnil ThoratNo ratings yet
- Hydrography and BathymetryDocument32 pagesHydrography and BathymetryArielle Skye PrzybyszNo ratings yet
- Waveguides & Microwave Devices: Lesson 1Document103 pagesWaveguides & Microwave Devices: Lesson 1Alas Mallari DonatoNo ratings yet
- B2017e-1 Proposal of Products For Machine ToolsDocument7 pagesB2017e-1 Proposal of Products For Machine ToolsElmer Lagua MalpasoNo ratings yet
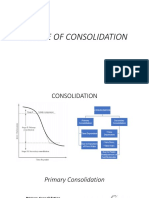
- Degree of Consolidation NewDocument13 pagesDegree of Consolidation NewrifaniNo ratings yet
- Effect of Cooling Rate and Finish Rolling Temperature On Structure and Strength of API 5LX70 Linepipe Steel PlateDocument11 pagesEffect of Cooling Rate and Finish Rolling Temperature On Structure and Strength of API 5LX70 Linepipe Steel Platek4dr0nNo ratings yet
- Wirring, Pipping Hitachi - ACDocument27 pagesWirring, Pipping Hitachi - ACHera WatiNo ratings yet
- LSA44.3 - Maintenance ManualDocument28 pagesLSA44.3 - Maintenance ManualMuhammad HaekalNo ratings yet
- Vibration Lab 2 Belt Friction Lab ReportDocument15 pagesVibration Lab 2 Belt Friction Lab ReportYann YeuNo ratings yet