You might also like
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (122)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (844)
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (590)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5810)
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1092)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (401)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (346)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (897)
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (540)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (822)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- Heredity - How A Protein Is Made Using DNA InformationDocument45 pagesHeredity - How A Protein Is Made Using DNA Informationkath chandriaNo ratings yet
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Prokaryotic and Eukaryotic CellsDocument2 pagesProkaryotic and Eukaryotic CellsSerena Adeline RiveraNo ratings yet
- Chapter17 Blood MariebDocument30 pagesChapter17 Blood MariebMario AndersonNo ratings yet
- Cell Analogy WorksheetDocument2 pagesCell Analogy Worksheetjasonchu1280% (5)
- Michele Maruccia, Giuseppe Giudice - Textbook of Plastic and Reconstructive Surgery - Basic Principles and New Perspectives-Springer (2022)Document534 pagesMichele Maruccia, Giuseppe Giudice - Textbook of Plastic and Reconstructive Surgery - Basic Principles and New Perspectives-Springer (2022)blume diaNo ratings yet
- Malnutrition in Stroke PatientDocument8 pagesMalnutrition in Stroke PatientFetria MelaniNo ratings yet
- Chap 4 Lactation and Human Milk Part 1Document12 pagesChap 4 Lactation and Human Milk Part 1Fetria MelaniNo ratings yet
- 006 Pathology MCQ ACEM Primary CellularDocument5 pages006 Pathology MCQ ACEM Primary CellularIrum RafeeqNo ratings yet
- ABMLI Sample Questions 000Document7 pagesABMLI Sample Questions 000samy100% (1)
- Cerebral Blood FlowDocument37 pagesCerebral Blood FlowFetria MelaniNo ratings yet
- Immunity and Aging1742-4933!5!15Document10 pagesImmunity and Aging1742-4933!5!15Fetria MelaniNo ratings yet
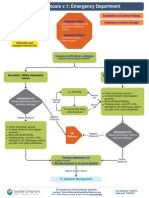
- Algorithm Pyloric StenosisDocument15 pagesAlgorithm Pyloric StenosisFetria MelaniNo ratings yet
- Nutrisi Ulseratif KolitisDocument1 pageNutrisi Ulseratif KolitisFetria MelaniNo ratings yet
- Full Liquid DietDocument1 pageFull Liquid DietFetria MelaniNo ratings yet
- The Anatomy of A Great Poster: Richard Gerkin, M.D. Bridget Stiegler, D.ODocument31 pagesThe Anatomy of A Great Poster: Richard Gerkin, M.D. Bridget Stiegler, D.OFetria MelaniNo ratings yet
- Health: Aradhna Kumar Bachelor of Health Science (Podiatry)Document10 pagesHealth: Aradhna Kumar Bachelor of Health Science (Podiatry)Fetria MelaniNo ratings yet
- Clear Liquid DietDocument3 pagesClear Liquid DietFetria Melani100% (1)
- Metabolisme Zat BesiDocument33 pagesMetabolisme Zat BesiFetria MelaniNo ratings yet
- Gene SilencingDocument24 pagesGene SilencingSUVODIP JANANo ratings yet
- Tugas Bu Poppy Cell - Cycle - Inhibition - and - Apoptotic - Induction - of - VDocument4 pagesTugas Bu Poppy Cell - Cycle - Inhibition - and - Apoptotic - Induction - of - VAndreNo ratings yet
- GB1 Q2 Week-3bDocument10 pagesGB1 Q2 Week-3bAnnejhel Mae PoralanNo ratings yet
- Extended Definition RevisedDocument6 pagesExtended Definition Revisedapi-491301139No ratings yet
- Cell Analogy Project 2021Document3 pagesCell Analogy Project 2021api-249551710No ratings yet
- Regulation of Peptidoglycan SynthesisDocument15 pagesRegulation of Peptidoglycan SynthesisVictor HugoNo ratings yet
- Kami Export - Enzyme - Check - in - Quiz - ANSWER KEYDocument3 pagesKami Export - Enzyme - Check - in - Quiz - ANSWER KEYNicholas UrrutiaNo ratings yet
- Class Xii Biology Chapter 6 - Molecular Basis of InheritanceDocument8 pagesClass Xii Biology Chapter 6 - Molecular Basis of InheritanceAkshit Sharma VIII Diasy Roll no 6No ratings yet
- Host Modulation Therapeutics in Periodontics: Role As An Adjunctive Periodontal TherapyDocument10 pagesHost Modulation Therapeutics in Periodontics: Role As An Adjunctive Periodontal Therapypaper kitaNo ratings yet
- Physical Activity and Cancer Risk. Actual Knowledge and Possible Biological MechanismsDocument11 pagesPhysical Activity and Cancer Risk. Actual Knowledge and Possible Biological MechanismsTiara SafitriNo ratings yet
- The Histone Code HypothesisDocument5 pagesThe Histone Code HypothesisBrian D. StrahlNo ratings yet
- Chimeric Peptidomimetic Antibiotic Efficiently Neutralizes Lipopolysaccharides (LPS) and Bacteria-Induced Activation of RAW MacrophagesDocument9 pagesChimeric Peptidomimetic Antibiotic Efficiently Neutralizes Lipopolysaccharides (LPS) and Bacteria-Induced Activation of RAW MacrophagesKATIA PAMELA VILLAVICENCIO SANCHEZNo ratings yet
- Inherited Giant Platelet Disorders PDFDocument15 pagesInherited Giant Platelet Disorders PDFpieterinpretoria391No ratings yet
- Stem CellsDocument18 pagesStem CellsJustine Mae CipresNo ratings yet
- Hemostasis: Written By: Fayzah Alshammari Date: 06-11-2021Document4 pagesHemostasis: Written By: Fayzah Alshammari Date: 06-11-2021fayzah alshammariNo ratings yet
- BIOL 3301 - Genetics Ch9A - Genetic Analysis and Mapping in BacteriaDocument36 pagesBIOL 3301 - Genetics Ch9A - Genetic Analysis and Mapping in BacteriaChuongNo ratings yet
- Ap Biology 2020 Practice Exam and Notes 3 FRQ Scoring GuidelinesDocument8 pagesAp Biology 2020 Practice Exam and Notes 3 FRQ Scoring GuidelinesEric ChiangNo ratings yet
- Cell Division Worksheet AnswersDocument3 pagesCell Division Worksheet AnswersDanaNo ratings yet
- Chapter 25 SkinDocument40 pagesChapter 25 SkinIsfahan MasulotNo ratings yet
- Muscle Tissue SummaryDocument2 pagesMuscle Tissue SummaryLindenScholesNo ratings yet
- Fatty Acid Oxidation & Ketone BodiesDocument30 pagesFatty Acid Oxidation & Ketone BodiesM.PRASAD NAIDUNo ratings yet
- ARS - NET Previous Questions - Agricultural Biotechnology Biology Exams 4 UDocument5 pagesARS - NET Previous Questions - Agricultural Biotechnology Biology Exams 4 UvalmikisatishNo ratings yet
- Part 1: Muscle Physiology: Skeletal MusclesDocument7 pagesPart 1: Muscle Physiology: Skeletal MusclesMyrtle Yvonne RagubNo ratings yet