You might also like
- Malnutrition DisordersDocument47 pagesMalnutrition DisordersAnnisa AstariNo ratings yet
- PreConception Care 4 Student VersionDocument38 pagesPreConception Care 4 Student VersionKajal SinghNo ratings yet
- PreConception CareDocument38 pagesPreConception CareMaya LubisNo ratings yet
- Thyroid Disease in PregnancyDocument70 pagesThyroid Disease in PregnancyRajeev Sood100% (1)
- Hypocalcemia in The NewbornDocument5 pagesHypocalcemia in The NewbornrenjithNo ratings yet
- Heart Diseases Diabetes Mellitus Substance Abuse Hiv/Aids RH Sensitization AnemiaDocument46 pagesHeart Diseases Diabetes Mellitus Substance Abuse Hiv/Aids RH Sensitization Anemiarevie67% (3)
- Pregestational ConditionsDocument66 pagesPregestational ConditionsEsvinch EsvinchNo ratings yet
- OB Drugs 2263Document43 pagesOB Drugs 2263Anthony Torres100% (5)
- Theme:: Early Gestosis. Late GestosisDocument17 pagesTheme:: Early Gestosis. Late GestosisPoisonIvyyyNo ratings yet
- Peads TDocument8 pagesPeads TAqsa ShNo ratings yet
- Wechatpediatrics+final NotesDocument5 pagesWechatpediatrics+final NotesPrasadNo ratings yet
- Pregestational ConditionDocument70 pagesPregestational ConditionCalista NovaNo ratings yet
- 109 Pregestational Nov2019Document68 pages109 Pregestational Nov2019Sean kevinNo ratings yet
- Breast Feeding + MalnutritionDocument33 pagesBreast Feeding + MalnutritionNight CrawlerNo ratings yet
- Infant of Diabetic MotherDocument29 pagesInfant of Diabetic MotherChro MANo ratings yet
- DOH ProgramsDocument66 pagesDOH ProgramsGina Sia SigatonNo ratings yet
- Revesion Nelson Notes 20Document205 pagesRevesion Nelson Notes 20Hanouf BakriNo ratings yet
- Infant Nutrition and Feeding RecommendationsDocument45 pagesInfant Nutrition and Feeding RecommendationsmanjitNo ratings yet
- NCM 109 PEDIA PPT 2 High Risk Infant A New Pre TermDocument72 pagesNCM 109 PEDIA PPT 2 High Risk Infant A New Pre TermJoyce EricaNo ratings yet
- Nutrition During Pregnancy Part2Document17 pagesNutrition During Pregnancy Part222bgu0727msNo ratings yet
- DM in PregnancyDocument11 pagesDM in Pregnancyميمونه عبد الرحيم مصطفىNo ratings yet
- Small Ruminant Nutritional DiseaseDocument11 pagesSmall Ruminant Nutritional DiseasePrince PrinceNo ratings yet
- Prognosis of common medical conditionsDocument1 pagePrognosis of common medical conditionsUpsetNo ratings yet
- Vaccination+ Metabolic Disorder.Document37 pagesVaccination+ Metabolic Disorder.hasanatiya41No ratings yet
- High Risk Nb.Document65 pagesHigh Risk Nb.Padmashree MannemNo ratings yet
- NCP For FT, SGADocument7 pagesNCP For FT, SGAJule Santoya80% (5)
- Diabetes PregnancyDocument65 pagesDiabetes PregnancyJohn Christopher LucesNo ratings yet
- Medical and Surgical Complicating PregnancyDocument70 pagesMedical and Surgical Complicating PregnancyGunaNo ratings yet
- Severe Acute Malnutrition and Fluid Management inDocument76 pagesSevere Acute Malnutrition and Fluid Management inBibsNo ratings yet
- Maternal ATIDocument6 pagesMaternal ATIGeorgeNo ratings yet
- Whooping CoughDocument72 pagesWhooping Coughwengie100% (1)
- Maternal Risk Factors and Fetal AssessmentDocument88 pagesMaternal Risk Factors and Fetal AssessmentLeofe CorregidorNo ratings yet
- Focus Vet. MagazineDocument6 pagesFocus Vet. MagazineAmurLeopardNo ratings yet
- Nursing Care of Children With Indian Childhood Cirrhosis, Wilsons Disesase, Reyes SyndromeDocument26 pagesNursing Care of Children With Indian Childhood Cirrhosis, Wilsons Disesase, Reyes SyndromeDivya Nair100% (2)
- C C C CDocument51 pagesC C C CpixiemedicNo ratings yet
- KIE Prognosis and Complications of Preterm BirthDocument3 pagesKIE Prognosis and Complications of Preterm BirthYolanda a.k.a YLNo ratings yet
- Life Span Health PromotionDocument67 pagesLife Span Health Promotion22bgu0727msNo ratings yet
- my paediatric notesDocument14 pagesmy paediatric notesTicky TomNo ratings yet
- AHD Dec 15 10 - Women's Issues in Neurology - YuDocument55 pagesAHD Dec 15 10 - Women's Issues in Neurology - YuoliveroslovelynNo ratings yet
- Obstetric ExamDocument194 pagesObstetric ExamHimmz100% (1)
- Formulations of Representative Drugs From Antibiotics, Antipyretics, Steroids, Injectables and VitaminsDocument4 pagesFormulations of Representative Drugs From Antibiotics, Antipyretics, Steroids, Injectables and VitaminsChandraprakash JangidNo ratings yet
- Maternal Problems in Pregnancy Briefing - 3amaliDocument14 pagesMaternal Problems in Pregnancy Briefing - 3amaliArsh KaiwanNo ratings yet
- Pregnancy and Lactation AnswersDocument8 pagesPregnancy and Lactation AnswersAoiNo ratings yet
- Pre Gestational DiseasesDocument86 pagesPre Gestational DiseasesChristian James MataNo ratings yet
- Gyn AsDocument2 pagesGyn AsJamal kenasaNo ratings yet
- Indian Childhood CirrhosisDocument10 pagesIndian Childhood CirrhosisMona Morris89% (9)
- Nutritional Support of Children With Chronic LiverDocument5 pagesNutritional Support of Children With Chronic LiverVianNo ratings yet
- Infant of Diabetic MotherDocument41 pagesInfant of Diabetic Mothermohdmaghyreh100% (8)
- Aspirin and Fish Oil Drug Study for Pregnancy CareDocument33 pagesAspirin and Fish Oil Drug Study for Pregnancy Careマリ ベルNo ratings yet
- Types of Childhood Diabetes and Treatment UpdatesDocument51 pagesTypes of Childhood Diabetes and Treatment UpdatesJulie Carnetion DNo ratings yet
- High Risk Infant-1Document5 pagesHigh Risk Infant-1MauZungNo ratings yet
- Small Ruminant Neonatology 2008Document19 pagesSmall Ruminant Neonatology 2008daniellopesrpNo ratings yet
- Pediatrics (Toronto Notes For PDA 2004)Document70 pagesPediatrics (Toronto Notes For PDA 2004)Worawut PhromwangNo ratings yet
- Malaria and Malnutrition PDFDocument1 pageMalaria and Malnutrition PDFadite_alifaNo ratings yet
- Nursing Care For High Risk NeonatesDocument13 pagesNursing Care For High Risk NeonatesWayne Patrick De LunaNo ratings yet
- Intra Uteri Growt RetradationDocument44 pagesIntra Uteri Growt RetradationFikriBelikeNo ratings yet
- Pyometra in Small Animals - Reproductive System - Veterinary Manual PDFDocument3 pagesPyometra in Small Animals - Reproductive System - Veterinary Manual PDFKirti JamwalNo ratings yet
- Naplex Complete Study Outline A Topic-Wise Approach DiabetesFrom EverandNaplex Complete Study Outline A Topic-Wise Approach DiabetesRating: 4 out of 5 stars4/5 (2)
- EXAM 1 2002 SampleDocument3 pagesEXAM 1 2002 SampleadamsamillionNo ratings yet
- Med Genetics Exam KeyDocument6 pagesMed Genetics Exam KeyadamsamillionNo ratings yet
- Final Genetics 2015Document33 pagesFinal Genetics 2015samNo ratings yet
- Genetics Key11111Document7 pagesGenetics Key11111adamsamillionNo ratings yet
- Step 1 Guide-2Document26 pagesStep 1 Guide-2João GalanteNo ratings yet
- Step 1 Experience - Amjad ElmashalaDocument4 pagesStep 1 Experience - Amjad ElmashalaadamsamillionNo ratings yet
- Beginner Body Weight Training Week 1 8 ScheduleDocument3 pagesBeginner Body Weight Training Week 1 8 SchedulesandeepNo ratings yet
- 002 Intermediate-Body-Weight-Training-Week-1-8-ScheduleDocument3 pages002 Intermediate-Body-Weight-Training-Week-1-8-ScheduleadamsamillionNo ratings yet
- 002 Advanced-Body-Weight-Training-Week-1-8-ScheduleDocument3 pages002 Advanced-Body-Weight-Training-Week-1-8-ScheduleadamsamillionNo ratings yet
- Neurology notes from 2020 cover key conceptsDocument20 pagesNeurology notes from 2020 cover key conceptsadamsamillionNo ratings yet
- Pediatric Health Supervision: Weight, Height, Head Circumference, and VaccinationsDocument57 pagesPediatric Health Supervision: Weight, Height, Head Circumference, and Vaccinationsadamsamillion100% (1)
- Obgyn UWISE Notes (And Master The Boards)Document8 pagesObgyn UWISE Notes (And Master The Boards)Laura Lopez Roca100% (5)
- UWorld Notes Step 2 CKDocument11 pagesUWorld Notes Step 2 CKSamah Khan50% (2)
- Pediatrics NotesDocument100 pagesPediatrics Notessanflash100% (1)
- 100 Concepts Anatomy PDFDocument38 pages100 Concepts Anatomy PDFTauseefUllah100% (1)
- The Roger Wyburn Mason M.D. Ph.D. Treatment For Rheumatoid DiseaseDocument13 pagesThe Roger Wyburn Mason M.D. Ph.D. Treatment For Rheumatoid DiseaseSteluța DrâmbuNo ratings yet
- Chylous Ascites 3Document3 pagesChylous Ascites 3Nur atikaNo ratings yet
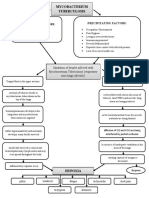
- Mycobacterium Tuberculosis: Precipitating Factors: Predisposing FactorsDocument1 pageMycobacterium Tuberculosis: Precipitating Factors: Predisposing FactorsYoko Mae Yano100% (1)
- Guidelines For The Design and Construction of Health Care FacilitiesDocument370 pagesGuidelines For The Design and Construction of Health Care Facilitiesmasoodae93% (15)
- Care of Older Adult (Rle)Document10 pagesCare of Older Adult (Rle)Antoinette PeleñaNo ratings yet
- Final CHN 2015Document37 pagesFinal CHN 2015MeeKo VideñaNo ratings yet
- Tuberculosis: Causes, Symptoms, Prevention & TreatmentDocument7 pagesTuberculosis: Causes, Symptoms, Prevention & TreatmentKiara Zarate ArribasplataNo ratings yet
- New Washington National Comprehensive High School Poblacion, New Washington, AklanDocument19 pagesNew Washington National Comprehensive High School Poblacion, New Washington, AklanJram MarjNo ratings yet
- Paediatrica Indonesiana: Fadilah Harahap, Ridwan M. Daulay, Muhammad Ali, Wisman Dalimunthe, Rini Savitri DaulayDocument11 pagesPaediatrica Indonesiana: Fadilah Harahap, Ridwan M. Daulay, Muhammad Ali, Wisman Dalimunthe, Rini Savitri DaulayMuhammadImamNo ratings yet
- Script Video About TBCDocument2 pagesScript Video About TBCMithaprnmsrNo ratings yet
- Ch 31 Asthma COPD TB ReviewDocument2 pagesCh 31 Asthma COPD TB Reviewremyde07No ratings yet
- Pre Employment Occupational Health FormDocument7 pagesPre Employment Occupational Health Formlinks2309No ratings yet
- Non-Communicable Diseases and Health Systems Reform in Low and Middle-Income Countries (WP13)Document24 pagesNon-Communicable Diseases and Health Systems Reform in Low and Middle-Income Countries (WP13)Nossal Institute for Global HealthNo ratings yet
- Pulmonary Tuberculosis Presenting with ARDSDocument5 pagesPulmonary Tuberculosis Presenting with ARDSCimol AgustinaNo ratings yet
- Tugas Rancangan Asuhan Keperawatan Dengan Integrasi Ebp DanDocument11 pagesTugas Rancangan Asuhan Keperawatan Dengan Integrasi Ebp DanInkha LepaNo ratings yet
- Melody Doria - Chapter 20 ActivityDocument3 pagesMelody Doria - Chapter 20 ActivityMelody MirandaNo ratings yet
- REVALIDA EXAM 200 Answers With RationaleDocument17 pagesREVALIDA EXAM 200 Answers With Rationalexaileenx100% (1)
- 1 Eqa SopDocument2 pages1 Eqa SopRajeev PareekNo ratings yet
- Mzu 010Document8 pagesMzu 010edi_wsNo ratings yet
- TNFα role in MTB infection control and tissue damageDocument3 pagesTNFα role in MTB infection control and tissue damagePinaufal Ahmad FNo ratings yet
- Diabetes Mellitus Increases The Risk of Active PDFDocument11 pagesDiabetes Mellitus Increases The Risk of Active PDFherry2swNo ratings yet
- WHO EMP IAU 2017.11 Eng PDFDocument48 pagesWHO EMP IAU 2017.11 Eng PDFKypexflyNo ratings yet
- Pathology of Musculoskeletal System: Bone Tumors and DiseasesDocument68 pagesPathology of Musculoskeletal System: Bone Tumors and DiseaseswilliamNo ratings yet
- 2007 AnswersDocument81 pages2007 AnswersTiffani Gutierrez100% (1)
- TUBERCULOSISDocument25 pagesTUBERCULOSISManuel Fatima Goncalves83% (6)
- Phase 2 Student HandbookDocument60 pagesPhase 2 Student HandbookCharlotte EeNo ratings yet
- Padma Priya Dars in I 2014Document12 pagesPadma Priya Dars in I 2014panji respatiNo ratings yet
- Vong 1Document10 pagesVong 1BlissKedNo ratings yet
- Saida, Syamsiar : Volume 7 Nomor 1 Bulan Oktober 2019 EISSN: 2443-0218Document9 pagesSaida, Syamsiar : Volume 7 Nomor 1 Bulan Oktober 2019 EISSN: 2443-0218neno fitriyani hasbieNo ratings yet
- The Six Killer DiseaseDocument26 pagesThe Six Killer DiseaseFrancis Luke63% (8)