You might also like
- Astm D 1418 PDFDocument3 pagesAstm D 1418 PDFseeralan_198667% (3)
- The Subversion of The Subject and The Dialectics of Desire in The Freudian UnconsciousDocument17 pagesThe Subversion of The Subject and The Dialectics of Desire in The Freudian UnconscioushegelhegelNo ratings yet
- Berg Violin Concerto ArticleDocument7 pagesBerg Violin Concerto ArticleSamNo ratings yet
- Berg Violin Concerto ArticleDocument7 pagesBerg Violin Concerto ArticleSamNo ratings yet
- International StandardsDocument93 pagesInternational StandardsakashdadNo ratings yet
- Analytical Assessment of The Mechanical Design of A Metal Getter Bed For Hydrogen StorageDocument15 pagesAnalytical Assessment of The Mechanical Design of A Metal Getter Bed For Hydrogen StorageRupshaBNo ratings yet
- H2S Removal from Biogas Using Iron-Chelated SolutionDocument8 pagesH2S Removal from Biogas Using Iron-Chelated SolutionsachinnandeNo ratings yet
- Review of Factors in Establishing an Effective MRFDocument10 pagesReview of Factors in Establishing an Effective MRFjohn paul pascuaNo ratings yet
- Climate BiodiversityDocument11 pagesClimate BiodiversitySamNo ratings yet
- BS 5075-2-1982 - Concrete Admixtures - Specification For Air Entraining AdmixturesDocument19 pagesBS 5075-2-1982 - Concrete Admixtures - Specification For Air Entraining AdmixturesRaviranjan kumarNo ratings yet
- Trombone Tips For ComposersDocument6 pagesTrombone Tips For Composersjosejolulu100% (1)
- Birmingham Pendulum ClockDocument27 pagesBirmingham Pendulum ClockJardel Santos de DeusNo ratings yet
- Rehabilitation of existing PCCP road from Daet to NagaDocument1 pageRehabilitation of existing PCCP road from Daet to NagaUzziah GonzalesNo ratings yet
- Burner Training Book PDFDocument32 pagesBurner Training Book PDFjns1606No ratings yet
- Aluminium Conductor Composite Core ACCCDocument5 pagesAluminium Conductor Composite Core ACCCAmlanshankar Deb BarmaNo ratings yet
- A330 21 L3Document341 pagesA330 21 L3Thanh Vinh Nguyen100% (3)
- ROCHEM RO-Wasserbehandlung GMBH PDFDocument117 pagesROCHEM RO-Wasserbehandlung GMBH PDFhdf1788% (8)
- Rural - Materials & ConstructionDocument12 pagesRural - Materials & ConstructionHaresh Babu PitchaimaniNo ratings yet
- Analisis Kinerja Pengolahan Air TificoDocument138 pagesAnalisis Kinerja Pengolahan Air TificoAndri Imam MunandarNo ratings yet
- Fischer-Tropsch (FT) ProcessDocument18 pagesFischer-Tropsch (FT) ProcessNur Ainie Baharudin100% (4)
- International Journal of Coal GeologyDocument12 pagesInternational Journal of Coal GeologyFirman FirmanNo ratings yet
- Hickman 1993Document4 pagesHickman 1993tieNo ratings yet
- Hydrogen CHEMHACKDocument7 pagesHydrogen CHEMHACKMonika BoranaNo ratings yet
- Research ArticleDocument7 pagesResearch ArticleAmer KasidehNo ratings yet
- Art 3A10.1134 2FS0023158412060067Document5 pagesArt 3A10.1134 2FS0023158412060067hoseiNo ratings yet
- Applied Catalysis B: EnvironmentalDocument6 pagesApplied Catalysis B: EnvironmentalMinh TrầnNo ratings yet
- Musashi 2000Document11 pagesMusashi 2000DanCosminNo ratings yet
- Erang PDFDocument8 pagesErang PDFSiddhartha paudelNo ratings yet
- CO2 Conversion To CO by Auto-Thermal Catalyst-Assisted Chemical LoopingDocument9 pagesCO2 Conversion To CO by Auto-Thermal Catalyst-Assisted Chemical LoopingNguyễn TuânNo ratings yet
- Diatomite As High Performance and Environmental Friendly Catalysts For Phenol Hydroxylation With H ODocument4 pagesDiatomite As High Performance and Environmental Friendly Catalysts For Phenol Hydroxylation With H ONguyễn DungNo ratings yet
- Generation of Hydroxyl RadicalsDocument9 pagesGeneration of Hydroxyl RadicalsRebeccaNo ratings yet
- Bartels 1982Document12 pagesBartels 1982giampieroNo ratings yet
- On the general mechanism of photocatalytic reduction of CO2Document10 pagesOn the general mechanism of photocatalytic reduction of CO2alejandro sifuentes clementeNo ratings yet
- Bierenstiel I In. - 2005 - Investigations Into The Selective Oxidation of VicDocument7 pagesBierenstiel I In. - 2005 - Investigations Into The Selective Oxidation of Vicantrios123No ratings yet
- Fenton and Photo FentonDocument10 pagesFenton and Photo FentonRohit ChauhanNo ratings yet
- Slimene DosimétrieDocument8 pagesSlimene DosimétrieSennaoui LaraNo ratings yet
- Degradation kinetics and mechanisms of phenol in photo-Fenton processDocument8 pagesDegradation kinetics and mechanisms of phenol in photo-Fenton processAhmed MohyNo ratings yet
- Selective Hydrogen Absorption From Gaseous Mixtures by BCC Ti-V AlloysDocument12 pagesSelective Hydrogen Absorption From Gaseous Mixtures by BCC Ti-V AlloyswarnoiseNo ratings yet
- Epoxide-Functionalization of Polyethyleneimine For Synthesis of Stable Carbon Dioxide Adsorbent in Temperature Swing AdsorptionDocument8 pagesEpoxide-Functionalization of Polyethyleneimine For Synthesis of Stable Carbon Dioxide Adsorbent in Temperature Swing AdsorptionFrida Octavia PurnomoNo ratings yet
- Musso 1963Document13 pagesMusso 1963AshleyNo ratings yet
- J Ijhydene 2020 03 096Document13 pagesJ Ijhydene 2020 03 096Wassachol SumarasinghaNo ratings yet
- Houas 2001Document13 pagesHouas 2001chem19111117No ratings yet
- 1 - Paper - Planta Piloto Absorcion H2S PDFDocument7 pages1 - Paper - Planta Piloto Absorcion H2S PDFSilas Calderon LuloNo ratings yet
- Effect of Temperature On The Solvent Extraction of Cobalt (II), Nickel (II), and Copper (II) Metal Ions by O-Diphenylamino Benzoic AcidDocument5 pagesEffect of Temperature On The Solvent Extraction of Cobalt (II), Nickel (II), and Copper (II) Metal Ions by O-Diphenylamino Benzoic AcidGustavo MamaniNo ratings yet
- Hydrogen Production From Water by Using Hydrogen Sulfide As Reducing Agent in Hydrothermal ReactionsDocument4 pagesHydrogen Production From Water by Using Hydrogen Sulfide As Reducing Agent in Hydrothermal ReactionsLukman HakimNo ratings yet
- AOPs for Remediating Recalcitrant Organic CompoundsDocument16 pagesAOPs for Remediating Recalcitrant Organic Compoundsshaik mohammed ArshadNo ratings yet
- 2005-Sonochemistry and Its DosimetryDocument6 pages2005-Sonochemistry and Its DosimetryOualid HamdaouiعععNo ratings yet
- GRE Minger 1982Document4 pagesGRE Minger 1982anhchangcodon88No ratings yet
- Chemical Reduction of Co To Dif Ferent Products During Photo Catalytic Reaction On Tio Under Diverse Conditions: An OverviewDocument10 pagesChemical Reduction of Co To Dif Ferent Products During Photo Catalytic Reaction On Tio Under Diverse Conditions: An Overviewaymanoh056No ratings yet
- Catalytic Conversion of Alkanes To Olefins by Carbon Dioxide Oxidative DehydrogenationA ReviewDocument14 pagesCatalytic Conversion of Alkanes To Olefins by Carbon Dioxide Oxidative DehydrogenationA ReviewBamrung SungnoenNo ratings yet
- S-Block For Jee AdvanceDocument38 pagesS-Block For Jee AdvanceSitabai JadhavNo ratings yet
- Selective Oxidation of Aldehydes To Carboxylic Acids With Sodium Chlorite-Hydrogen PeroxideDocument3 pagesSelective Oxidation of Aldehydes To Carboxylic Acids With Sodium Chlorite-Hydrogen PeroxidejavasoloNo ratings yet
- Photocatalytic Decolorization of Bismarck Brown RDocument170 pagesPhotocatalytic Decolorization of Bismarck Brown Rgeorge.ejaam8975No ratings yet
- 07 Hydroformylation 2023Document48 pages07 Hydroformylation 2023stadnichuk.timofeyNo ratings yet
- Tetrahedron Letters: Graziano Baccolini, Carla Boga, Camilla Delpivo, Gabriele MichelettiDocument5 pagesTetrahedron Letters: Graziano Baccolini, Carla Boga, Camilla Delpivo, Gabriele MichelettiRiyadh RayhandhiaNo ratings yet
- Homoacetogenesis As The Alternative Pathway For HDocument7 pagesHomoacetogenesis As The Alternative Pathway For HProfessor Douglas TorresNo ratings yet
- Journal of Industrial and Engineering Chemistry: Ines Nitoi, Tatiana Oncescu, Petruta OanceaDocument5 pagesJournal of Industrial and Engineering Chemistry: Ines Nitoi, Tatiana Oncescu, Petruta OanceaAndrea SilvaNo ratings yet
- Kinetics and Adsorption Studies of Lead (II) Onto Activated Carbon Using Low Cost AdsorbentsDocument8 pagesKinetics and Adsorption Studies of Lead (II) Onto Activated Carbon Using Low Cost AdsorbentsUmi LatifahNo ratings yet
- Journal of Environmental Chemical Engineering: SciencedirectDocument7 pagesJournal of Environmental Chemical Engineering: SciencedirectMaria Camila VieraNo ratings yet
- Photocatalytic Degradation Pathway of Methylene Blue in WaterDocument14 pagesPhotocatalytic Degradation Pathway of Methylene Blue in WaterRIVALDO MARSEL TNo ratings yet
- PyridinesDocument14 pagesPyridineskim haksongNo ratings yet
- MoradiDocument6 pagesMoradiDarian HerascuNo ratings yet
- Effect of Enviromental Conditions On PB (II) Adsorption Onto MnO2Document7 pagesEffect of Enviromental Conditions On PB (II) Adsorption Onto MnO2Echa EksantiNo ratings yet
- Production and Decomposition Dynamics of Hydrogen Peroxide in FreshwaterDocument6 pagesProduction and Decomposition Dynamics of Hydrogen Peroxide in Freshwateralex samNo ratings yet
- Direct Synthesis of Formic Acid From Carbon Dioxide by Hydrogenation in Acidic MediaDocument8 pagesDirect Synthesis of Formic Acid From Carbon Dioxide by Hydrogenation in Acidic MediaWilly ChandraNo ratings yet
- Efficient hydrogen production using nuclear heat from an HTGR via the iodine-sulfur processDocument7 pagesEfficient hydrogen production using nuclear heat from an HTGR via the iodine-sulfur processshameem siddiqueNo ratings yet
- 1 s2.0 S101060300400067X MainDocument6 pages1 s2.0 S101060300400067X MainL ZhangNo ratings yet
- Partial Oxidation of Methane To Synthesis Gas Using Carbon DioxideDocument2 pagesPartial Oxidation of Methane To Synthesis Gas Using Carbon DioxideAniket KaushalNo ratings yet
- Ccxxviii: .-Natural Glucosides. The Position of The Biose Residue in HesperidinDocument6 pagesCcxxviii: .-Natural Glucosides. The Position of The Biose Residue in HesperidinJeremiaNo ratings yet
- Vel Leitner 1996Document7 pagesVel Leitner 1996Markus MeierNo ratings yet
- Steam Reforming of Ethanol Over Metal-Oxide-Promoted PtSiO2 Catalysts Effects ofDocument4 pagesSteam Reforming of Ethanol Over Metal-Oxide-Promoted PtSiO2 Catalysts Effects ofDana MateiNo ratings yet
- Thelmechanisms: of Reductive Carboxylation ReactionsDocument8 pagesThelmechanisms: of Reductive Carboxylation ReactionsRaymond LaBoyNo ratings yet
- Selective Monoetherification of 1,4-Hydroquinone Promoted by NanoDocument6 pagesSelective Monoetherification of 1,4-Hydroquinone Promoted by NanoNuteLLa Gaming (EFL)No ratings yet
- CuO Reduction Mechanism PDFDocument9 pagesCuO Reduction Mechanism PDFAnna OlszewskaNo ratings yet
- Hydrogen and its compounds: key properties and reactionsDocument5 pagesHydrogen and its compounds: key properties and reactionsGagan NdNo ratings yet
- Organometallic Chemistry: Plenary Lectures Presented at the Fourth International Conference on Organometallic ChemistryFrom EverandOrganometallic Chemistry: Plenary Lectures Presented at the Fourth International Conference on Organometallic ChemistryF. G. A. StoneNo ratings yet
- Progress in Reaction Kinetics: Volume 6From EverandProgress in Reaction Kinetics: Volume 6K. R. JenningsNo ratings yet
- 35ApplB - P Dopedg C3N4Document13 pages35ApplB - P Dopedg C3N4SamNo ratings yet
- Why Plants Need Nutrients PDFDocument1 pageWhy Plants Need Nutrients PDFSamNo ratings yet
- The Need To Go Green: Patriots and Principal Prepare To Part WaysDocument24 pagesThe Need To Go Green: Patriots and Principal Prepare To Part WaysSamNo ratings yet
- Scienceand Justice 562016223230Document9 pagesScienceand Justice 562016223230SamNo ratings yet
- 2017 MISP Flyer With College List 9.18.17 PDFDocument2 pages2017 MISP Flyer With College List 9.18.17 PDFSamNo ratings yet
- Lab Preparing Solutions - NaClDocument2 pagesLab Preparing Solutions - NaClSamNo ratings yet
- WS 2 ConcentrationDocument3 pagesWS 2 ConcentrationSamNo ratings yet
- Microdosing Peganum Harmala: January 2018Document5 pagesMicrodosing Peganum Harmala: January 2018SamNo ratings yet
- 1977c XV UnsatTribromidePineneDocument5 pages1977c XV UnsatTribromidePineneSamNo ratings yet
- Robert Frosts Out OutDocument3 pagesRobert Frosts Out OutSamNo ratings yet
- Electronegativity Bond Polarity: Electronegativity of An ElementDocument12 pagesElectronegativity Bond Polarity: Electronegativity of An ElementSamNo ratings yet
- Religions: The Laughter of Fools: The Relevance of Wisdom in Today's WorldDocument10 pagesReligions: The Laughter of Fools: The Relevance of Wisdom in Today's WorldSamNo ratings yet
- Turmeric A Herbal and Traditional MedicineDocument24 pagesTurmeric A Herbal and Traditional MedicineSamNo ratings yet
- ANGUS NE Nitroethane SDSDocument18 pagesANGUS NE Nitroethane SDSSamNo ratings yet
- Prokop HumanFearsDocument6 pagesProkop HumanFearsSamNo ratings yet
- HuayanNumismaticsasMetaphysics OFFICIAL PDFDocument25 pagesHuayanNumismaticsasMetaphysics OFFICIAL PDFSamNo ratings yet
- Lapping 2008 Ethics 69Document20 pagesLapping 2008 Ethics 69SamNo ratings yet
- What Made Gandhis Non Violent Movement WorkDocument15 pagesWhat Made Gandhis Non Violent Movement WorkSamNo ratings yet
- CrossProductAnd 2DGeometry PDFDocument1 pageCrossProductAnd 2DGeometry PDFSamNo ratings yet
- Template and Design LibraryDocument1 pageTemplate and Design LibrarySamNo ratings yet
- SQ HCU1 - AnswersDocument4 pagesSQ HCU1 - AnswersSamNo ratings yet
- Writing For ScholarshipsDocument2 pagesWriting For ScholarshipsSamNo ratings yet
- The Voice of Tommy WiseauDocument1 pageThe Voice of Tommy WiseauSamNo ratings yet
- How To Make Strong TeaDocument2 pagesHow To Make Strong TeaSamNo ratings yet
- Raymond Hand Pallet Trucks Product Reference GuideDocument2 pagesRaymond Hand Pallet Trucks Product Reference Guidemirzaadilbeg9No ratings yet
- Cera Flex - 80Document2 pagesCera Flex - 80harish GiriNo ratings yet
- 1.9 Electrostatics (I)Document10 pages1.9 Electrostatics (I)Jimmy ReeceNo ratings yet
- How desliming cyclones remove ash and fines from coalDocument3 pagesHow desliming cyclones remove ash and fines from coalRodrigo GarcíaNo ratings yet
- Expandable Mandrel Enquiry FormDocument3 pagesExpandable Mandrel Enquiry Formali-masoodNo ratings yet
- Chapter 10 - Product IdentificationDocument6 pagesChapter 10 - Product Identificationjtpml100% (1)
- Lithonia Outdoor KK Series Area Post Top Brochure 1-89Document10 pagesLithonia Outdoor KK Series Area Post Top Brochure 1-89Alan MastersNo ratings yet
- Electrical Symbols and Designations: Información de ServicioDocument4 pagesElectrical Symbols and Designations: Información de ServicioCEVegaONo ratings yet
- Non-Metallic Concrete Floor HardenerDocument4 pagesNon-Metallic Concrete Floor Hardener231340No ratings yet
- Falsework For Concrete Structures - Guidelines: Indian StandardDocument24 pagesFalsework For Concrete Structures - Guidelines: Indian StandardLivinston JosephNo ratings yet
- Top 100 Concrete Technology Interview Questions and AnswersDocument49 pagesTop 100 Concrete Technology Interview Questions and Answers210140106013.zaheeransariNo ratings yet
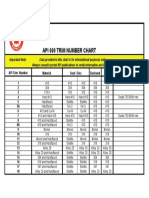
- API 600 Trim Number Chart..Document1 pageAPI 600 Trim Number Chart..francisco abarcaNo ratings yet
- ToolsDocument7 pagesToolsMallari, Matthew G.No ratings yet
- Aqeuous CorrosionDocument11 pagesAqeuous Corrosionthankz4venomNo ratings yet
- Larsen & Toubro Limited, Construction.: WOM Bill Annexure - EC578BIL0000026 DT:09 Apr 2020Document4 pagesLarsen & Toubro Limited, Construction.: WOM Bill Annexure - EC578BIL0000026 DT:09 Apr 2020Kannan GnanaprakasamNo ratings yet
- Alloy Vs CompositeDocument1 pageAlloy Vs CompositeankushNo ratings yet
- STL DatasheetDocument16 pagesSTL DatasheetCarlos Miguel Barrena TorresNo ratings yet