You might also like
- Shoe Dog: A Memoir by the Creator of NikeFrom EverandShoe Dog: A Memoir by the Creator of NikeRating: 4.5 out of 5 stars4.5/5 (537)
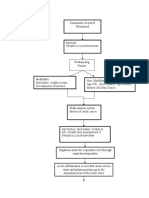
- Peripheral Artery Disease: Nikhil Vaishnav M.Sc. (Nursing)Document70 pagesPeripheral Artery Disease: Nikhil Vaishnav M.Sc. (Nursing)Dimpal ChoudharyNo ratings yet
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeFrom EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeRating: 4 out of 5 stars4/5 (5794)
- Pathophysiology of Pleural EffusionDocument1 pagePathophysiology of Pleural EffusionDimpal ChoudharyNo ratings yet
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceFrom EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceRating: 4 out of 5 stars4/5 (895)
- Streptococcus Pneumonae: Pathophysiology of CopdDocument1 pageStreptococcus Pneumonae: Pathophysiology of CopdDimpal ChoudharyNo ratings yet
- The Yellow House: A Memoir (2019 National Book Award Winner)From EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Rating: 4 out of 5 stars4/5 (98)
- Pathophysiology of PneumoniaDocument4 pagesPathophysiology of PneumoniaDimpal ChoudharyNo ratings yet
- Grit: The Power of Passion and PerseveranceFrom EverandGrit: The Power of Passion and PerseveranceRating: 4 out of 5 stars4/5 (588)
- Pulmonary FibrosisDocument4 pagesPulmonary FibrosisDimpal Choudhary100% (2)
- The Little Book of Hygge: Danish Secrets to Happy LivingFrom EverandThe Little Book of Hygge: Danish Secrets to Happy LivingRating: 3.5 out of 5 stars3.5/5 (400)
- Nursing Assessment Nursing Diagnosis Goal Planning Rationale Implementation Evaluation Subjective DataDocument5 pagesNursing Assessment Nursing Diagnosis Goal Planning Rationale Implementation Evaluation Subjective DataDimpal Choudhary100% (1)
- The Emperor of All Maladies: A Biography of CancerFrom EverandThe Emperor of All Maladies: A Biography of CancerRating: 4.5 out of 5 stars4.5/5 (271)
- Nursing Assessment Nursing Diagnosis Goal Planning Rationale Implementation Evaluation Subjective DataDocument4 pagesNursing Assessment Nursing Diagnosis Goal Planning Rationale Implementation Evaluation Subjective DataDimpal ChoudharyNo ratings yet
- Never Split the Difference: Negotiating As If Your Life Depended On ItFrom EverandNever Split the Difference: Negotiating As If Your Life Depended On ItRating: 4.5 out of 5 stars4.5/5 (838)
- Bladder IrrigationDocument5 pagesBladder IrrigationDimpal ChoudharyNo ratings yet
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyFrom EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyRating: 3.5 out of 5 stars3.5/5 (2259)
- Lumber Puncture ProcedureDocument8 pagesLumber Puncture ProcedureDimpal ChoudharyNo ratings yet
- On Fire: The (Burning) Case for a Green New DealFrom EverandOn Fire: The (Burning) Case for a Green New DealRating: 4 out of 5 stars4/5 (74)
- Procedure On Lumber PunctureDocument8 pagesProcedure On Lumber PunctureDimpal ChoudharyNo ratings yet
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureFrom EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureRating: 4.5 out of 5 stars4.5/5 (474)
- Congenital Heart DiseaseDocument82 pagesCongenital Heart DiseaseDimpal ChoudharyNo ratings yet
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryFrom EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryRating: 3.5 out of 5 stars3.5/5 (231)
- Uninodular Goitre: One Thyroid Nodule Can Be Either Inactive, or Active (Toxic)Document6 pagesUninodular Goitre: One Thyroid Nodule Can Be Either Inactive, or Active (Toxic)Dimpal ChoudharyNo ratings yet
- Team of Rivals: The Political Genius of Abraham LincolnFrom EverandTeam of Rivals: The Political Genius of Abraham LincolnRating: 4.5 out of 5 stars4.5/5 (234)
- Nursing EducationDocument19 pagesNursing EducationDimpal ChoudharyNo ratings yet
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaFrom EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaRating: 4.5 out of 5 stars4.5/5 (266)
- Procedure On Chest PhysiotherapyDocument13 pagesProcedure On Chest PhysiotherapyDimpal ChoudharyNo ratings yet
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersFrom EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersRating: 4.5 out of 5 stars4.5/5 (345)
- Preparation OF Group Work: Netaji Subhash College of NursingDocument7 pagesPreparation OF Group Work: Netaji Subhash College of NursingDimpal ChoudharyNo ratings yet
- Self-Directed LearningDocument18 pagesSelf-Directed LearningDimpal ChoudharyNo ratings yet
- The Unwinding: An Inner History of the New AmericaFrom EverandThe Unwinding: An Inner History of the New AmericaRating: 4 out of 5 stars4/5 (45)
- Polycythemia Vera: What Are The Symptoms of Polycythemia?Document10 pagesPolycythemia Vera: What Are The Symptoms of Polycythemia?Dimpal ChoudharyNo ratings yet
- Introduction-:: Time Specific Objective Content Av AidsDocument7 pagesIntroduction-:: Time Specific Objective Content Av AidsDimpal ChoudharyNo ratings yet
- It Is of Two Types Natural Disaster Man - Made DisasterDocument4 pagesIt Is of Two Types Natural Disaster Man - Made DisasterDimpal ChoudharyNo ratings yet
- Care Plan On PneumoniaDocument22 pagesCare Plan On PneumoniaDimpal Choudhary100% (3)
- 14-1 and 14-2 NotesDocument12 pages14-1 and 14-2 NotesVictoria ShinNo ratings yet
- A Neural Model For The Prediction of Pathogenic Genomic Variants in Mendelian DiseasesDocument7 pagesA Neural Model For The Prediction of Pathogenic Genomic Variants in Mendelian DiseasesGiorgio ValentiniNo ratings yet
- Premarital Screening ProgramDocument6 pagesPremarital Screening Programbaashii4No ratings yet
- Non Mendelian GeneticsDocument58 pagesNon Mendelian GeneticsRey Pineda100% (1)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreFrom EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreRating: 4 out of 5 stars4/5 (1090)
- AMNIOCENTESISDocument11 pagesAMNIOCENTESISaanya7febNo ratings yet
- Intellectual DisabilityDocument21 pagesIntellectual DisabilityNerezza SeleuinNo ratings yet
- Lynch SyndromeDocument6 pagesLynch SyndromeduminduNo ratings yet
- CHAPTER 1 - Uses of Biochemical Data - 2014 - Clinical Biochemistry MetabolicDocument5 pagesCHAPTER 1 - Uses of Biochemical Data - 2014 - Clinical Biochemistry Metabolicnour achkarNo ratings yet
- Physical Disability LearnersDocument25 pagesPhysical Disability LearnersChristian Luis De GuzmanNo ratings yet
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)From EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Rating: 4.5 out of 5 stars4.5/5 (121)
- Hemophilia ADocument20 pagesHemophilia Adiaharmayukti100% (1)
- Role of Genetics in Growth and DevelopmentDocument29 pagesRole of Genetics in Growth and DevelopmentEsha AslamNo ratings yet
- LSM1102 - Pract 7 Genetic Pedigree Analysis ExerciseDocument6 pagesLSM1102 - Pract 7 Genetic Pedigree Analysis Exercisegivena2ndchanceNo ratings yet
- Educational Psychology by John Santrock - 5e, TEST BANK 007337878XDocument52 pagesEducational Psychology by John Santrock - 5e, TEST BANK 007337878XjksnmmmNo ratings yet
- Reading Test - 19 Part - A: TIME: 15 MinutesDocument17 pagesReading Test - 19 Part - A: TIME: 15 MinutesKelvin KanengoniNo ratings yet
- Post TestDocument1 pagePost TestRosemarie R. ReyesNo ratings yet
- Understanding SmaDocument40 pagesUnderstanding SmaThanos ZihnalisNo ratings yet
- Her Body and Other Parties: StoriesFrom EverandHer Body and Other Parties: StoriesRating: 4 out of 5 stars4/5 (821)
- Pediatric Nursing Week 1Document3 pagesPediatric Nursing Week 1Jonah MaasinNo ratings yet
- DNA Diagnosis of Genetic DiseasesDocument40 pagesDNA Diagnosis of Genetic DiseasesSharanabasappa DurgNo ratings yet
- WWW - Natures.Ir: More Free Usmle, Mccee, Mcqe and Amq FlashcardsDocument54 pagesWWW - Natures.Ir: More Free Usmle, Mccee, Mcqe and Amq FlashcardsNixon GoyalNo ratings yet
- Tests All Topics PDFDocument215 pagesTests All Topics PDFYashwanth vNo ratings yet
- Causes of HypotoniaDocument2 pagesCauses of HypotoniaPutri Rahmi MaharaniNo ratings yet
- Types of Collagen and Associated Disorders: Osteogenesis ImperfectaDocument2 pagesTypes of Collagen and Associated Disorders: Osteogenesis ImperfectaAnjana J PrathapNo ratings yet
- 2023-05-12T08 - 05 - 49.1880033Z - Esb - Medical - Profile 2Document8 pages2023-05-12T08 - 05 - 49.1880033Z - Esb - Medical - Profile 2claudiais231966No ratings yet
- Progeria Case StudyDocument12 pagesProgeria Case StudyChristein Roger Dondoyano GamaleNo ratings yet
- Congenital Fibrosis of The Extra-Ocular Muscles Syndrome Review ArticleDocument5 pagesCongenital Fibrosis of The Extra-Ocular Muscles Syndrome Review Articleanon_703701023No ratings yet
- Bilateral Vestibulopathy: Diagnostic CriteriaDocument22 pagesBilateral Vestibulopathy: Diagnostic CriteriaHerminaElenaNo ratings yet
- Genetic TestingDocument112 pagesGenetic Testingprabhdeep kaurNo ratings yet
- M-1 MCQDocument88 pagesM-1 MCQFrt TrfNo ratings yet
- 2-Hyperprolinemia As A Clue in The Diagnosis of A Patientwith Psychiatric ManifestationsDocument3 pages2-Hyperprolinemia As A Clue in The Diagnosis of A Patientwith Psychiatric ManifestationsAli Emre KöseNo ratings yet
- Genetic CounsellingDocument52 pagesGenetic CounsellingVijetha RaiNo ratings yet
- Love Life: How to Raise Your Standards, Find Your Person, and Live Happily (No Matter What)From EverandLove Life: How to Raise Your Standards, Find Your Person, and Live Happily (No Matter What)Rating: 3 out of 5 stars3/5 (1)
- Summary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedFrom EverandSummary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedRating: 4.5 out of 5 stars4.5/5 (82)
- ADHD is Awesome: A Guide to (Mostly) Thriving with ADHDFrom EverandADHD is Awesome: A Guide to (Mostly) Thriving with ADHDRating: 5 out of 5 stars5/5 (3)
- LIT: Life Ignition Tools: Use Nature's Playbook to Energize Your Brain, Spark Ideas, and Ignite ActionFrom EverandLIT: Life Ignition Tools: Use Nature's Playbook to Energize Your Brain, Spark Ideas, and Ignite ActionRating: 4 out of 5 stars4/5 (404)